

Eval: a software package for analysis of genome annotations
- PMID: 14565849
- PMCID: PMC270064
- DOI: 10.1186/1471-2105-4-50
Eval: a software package for analysis of genome annotations
Abstract
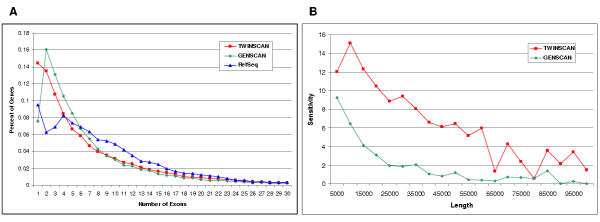
Summary: Eval is a flexible tool for analyzing the performance of gene annotation systems. It provides summaries and graphical distributions for many descriptive statistics about any set of annotations, regardless of their source. It also compares sets of predictions to standard annotations and to one another. Input is in the standard Gene Transfer Format (GTF). Eval can be run interactively or via the command line, in which case output options include easily parsable tab-delimited files.
Availability: To obtain the module package with documentation, go to http://genes.cse.wustl.edu/ and follow links for Resources, then Software. Please contact brent@cse.wustl.edu
Figures
References
-
- Korf I, Flicek P, Duan D, Brent MR. Integrating genomic homology into gene structure prediction. Bioinformatics. 2001;17 Suppl 1:S140–8. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
