

The genome of Nanoarchaeum equitans: insights into early archaeal evolution and derived parasitism
- PMID: 14566062
- PMCID: PMC240731
- DOI: 10.1073/pnas.1735403100
The genome of Nanoarchaeum equitans: insights into early archaeal evolution and derived parasitism
Abstract
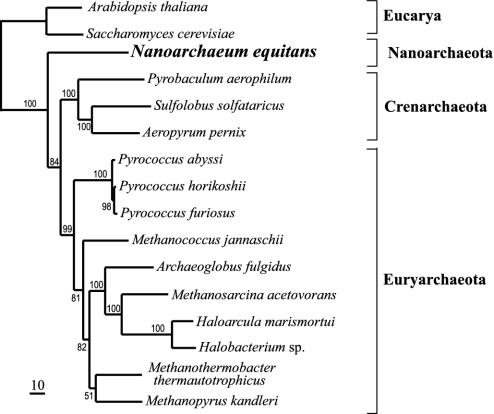
The hyperthermophile Nanoarchaeum equitans is an obligate symbiont growing in coculture with the crenarchaeon Ignicoccus. Ribosomal protein and rRNA-based phylogenies place its branching point early in the archaeal lineage, representing the new archaeal kingdom Nanoarchaeota. The N. equitans genome (490,885 base pairs) encodes the machinery for information processing and repair, but lacks genes for lipid, cofactor, amino acid, or nucleotide biosyntheses. It is the smallest microbial genome sequenced to date, and also one of the most compact, with 95% of the DNA predicted to encode proteins or stable RNAs. Its limited biosynthetic and catabolic capacity indicates that N. equitans' symbiotic relationship to Ignicoccus is parasitic, making it the only known archaeal parasite. Unlike the small genomes of bacterial parasites that are undergoing reductive evolution, N. equitans has few pseudogenes or extensive regions of noncoding DNA. This organism represents a basal archaeal lineage and has a highly reduced genome.
Figures
References
-
- Huber, H., Hohn, M. J., Rachel, R., Fuchs, T., Wimmer, V. C. & Stetter, K. O. (2002) Nature 417, 63-67. - PubMed
-
- Boucher, Y. & Doolittle, W. F. (2002) Nature 417, 27-28. - PubMed
-
- Huber, H., Hohn, M. J., Stetter, K. O. & Rachel, R. (2003) Res. Microbiol. 154, 165-171. - PubMed
-
- Silva, F. J., Latorre, A. & Moya, A. (2001) Trends Genet. 17, 615-618. - PubMed
-
- Ochman, H., Lawrence, J. G. & Groisman, E. A. (2000) Nature 405, 299-304. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
