

Critical role of the complement system in group B streptococcus-induced tumor necrosis factor alpha release
- PMID: 14573654
- PMCID: PMC219573
- DOI: 10.1128/IAI.71.11.6344-6353.2003
Critical role of the complement system in group B streptococcus-induced tumor necrosis factor alpha release
Abstract
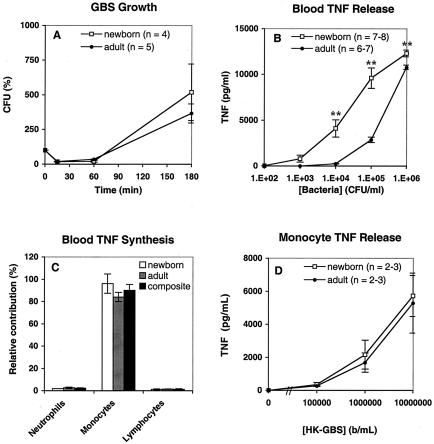
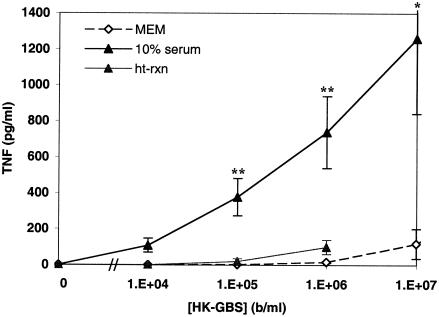
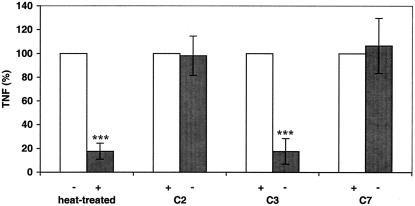
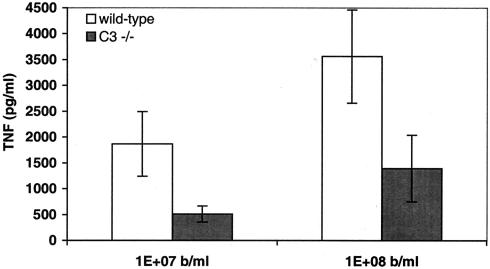
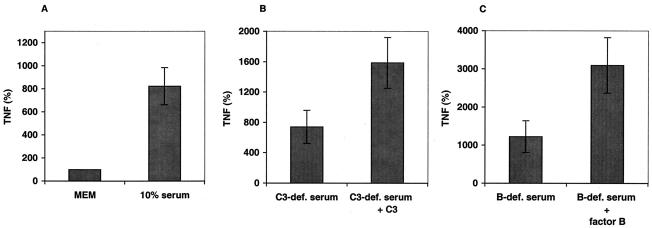
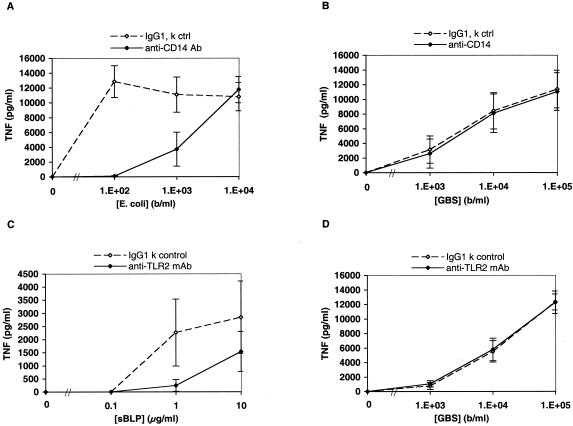
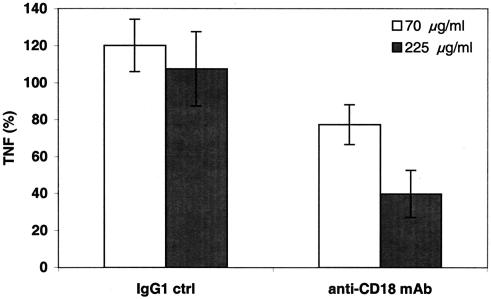
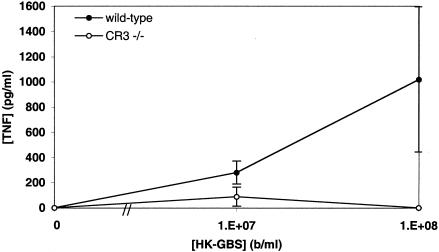
Group B Streptococcus (GBS) is a major cause of newborn sepsis and meningitis and induces systemic release of tumor necrosis factor alpha (TNF-alpha), believed to play a role in morbidity and mortality. While previous studies have shown that GBS can induce TNF-alpha release from monocytes and macrophages, little is known about the potential modulating effect of plasma or serum on GBS-induced TNF-alpha release, and there are conflicting reports as to the host receptors involved. In a human whole-blood assay system, GBS type III COH-1 potently induced substantial monocyte TNF-alpha release in adult peripheral blood and, due to a higher concentration of monocytes, 10-fold-greater TNF-alpha release in newborn cord blood. Remarkably, GBS-induced TNF-alpha release from human monocytes was enhanced approximately 1000-fold by heat-labile serum components. Experiments employing C2-, C3-, or C7-depleted serum demonstrated that C3 activation via the alternative pathway is crucial for potent GBS-induced TNF-alpha release. Accordingly, whole blood from C3-deficient mice demonstrated significantly reduced GBS-induced TNF-alpha release. Preincubation with human serum enhanced the TNF-alpha-inducing activity of GBS in a C3- and factor B-dependent manner, implying deposition of complement components via the alternative pathway. GBS-induced TNF-alpha release was inhibited by monoclonal antibodies directed against each of the components of CR3 and CR4: the common integrin beta subunit CD18 and the alpha subunits CD11b (of CR3) and CD11c (of CR4). Blood derived from CR3 (CD11b/CD18)-deficient mice demonstrated a markedly diminished TNF-alpha response to GBS. We conclude that the ability of plasma and serum to greatly amplify GBS-induced TNF-alpha release reflects the activity of the alternative complement pathway that deposits fragments on GBS and thereby enhances CR3- and CR4-mediated monocyte activation.
Figures
References
-
- Aderem, A., and R. J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature 406:782-787. - PubMed
-
- Aliprantis, A. O., R. B. Yang, M. R. Mark, S. Suggett, B. Devaux, J. D. Radolf, G. R. Klimpel, P. Godowski, and A. Zychlinsky. 1999. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285:736-739. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
