

Protein-truncating mutations in ASPM cause variable reduction in brain size
- PMID: 14574646
- PMCID: PMC1180496
- DOI: 10.1086/379085
Protein-truncating mutations in ASPM cause variable reduction in brain size
Abstract
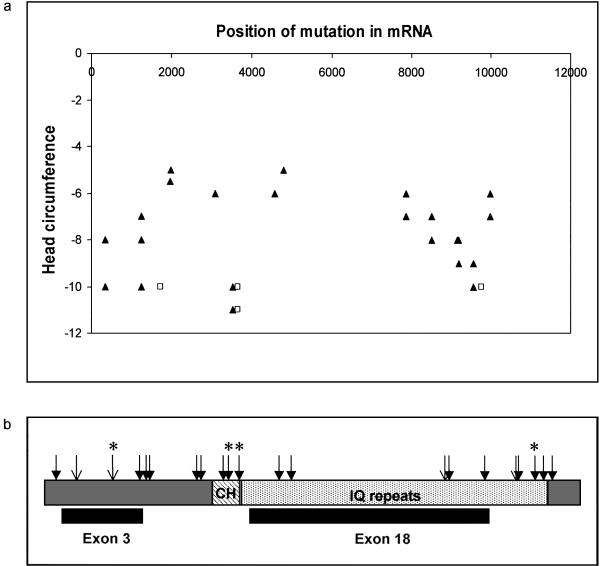
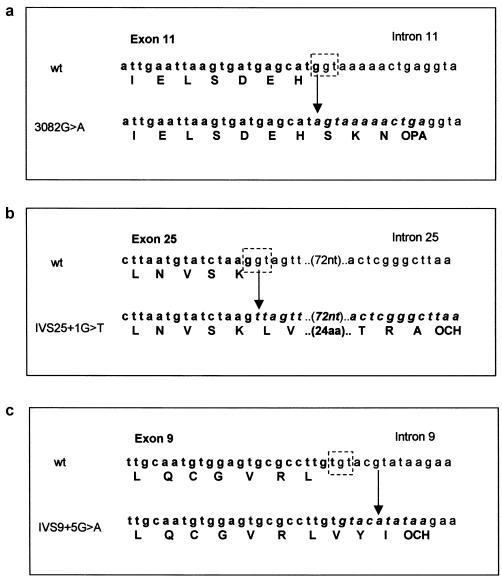
Mutations in the ASPM gene at the MCPH5 locus are expected to be the most common cause of human autosomal recessive primary microcephaly (MCPH), a condition in which there is a failure of normal fetal brain development, resulting in congenital microcephaly and mental retardation. We have performed the first comprehensive mutation screen of the 10.4-kb ASPM gene, identifying all 19 mutations in a cohort of 23 consanguineous families. Mutations occurred throughout the ASPM gene and were all predicted to be protein truncating. Phenotypic variation in the 51 affected individuals occurred in the degree of microcephaly (5-11 SDs below normal) and of mental retardation (mild to severe) but appeared independent of mutation position.
Figures

References
Electronic-Database Information
-
- Berkeley Drosophila Genome Project, http://www.fruitfly.org/seq_tools/splice.html (for splice prediction)
-
- INFOBIOGEN, http://www.infobiogen.fr/services/analyseq/cgi-bin/traduc_in.pl (for translation of nucleotide sequence to amino acids)
-
- National Center for Biotechnology Information (NCBI), Entrez search engine, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Nucleotide (for ASPM)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/ (for MCPH [MIM 251200] and ASPM [MIM 650481])
References
-
- Aicardi J (1998) Malformations of the central nervous system. In: Diseases of the nervous system in childhood, 2nd ed. Mac Keith, London, pp 90–91
-
- Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, Walsh CA, Woods CG (2002) ASPM is a major determinant of cerebral cortical size. Nat Genet 32:316–320 - PubMed
-
- Bundey S (1997) Abnormal Mental Development. In: Rimoin DL, Connor JM, Pyeritz RE (eds) Emery and Rimoin’s principles and practice of medical genetics, 3rd ed. Churchill Livingstone, New York, pp 730–731
-
- Craig R, Norbury C (1998) The novel murine calmodulin-binding protein Sha1 disrupts mitotic spindle and replication checkpoint functions in fission yeast. J Cell Sci 111:3609–3619 - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
