

Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor
- PMID: 14576166
- PMCID: PMC1360222
- DOI: 10.1074/jbc.M306248200
Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor
Abstract
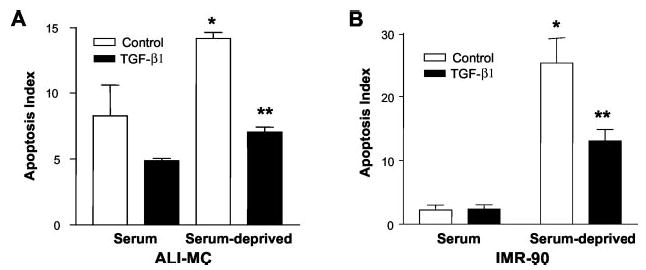
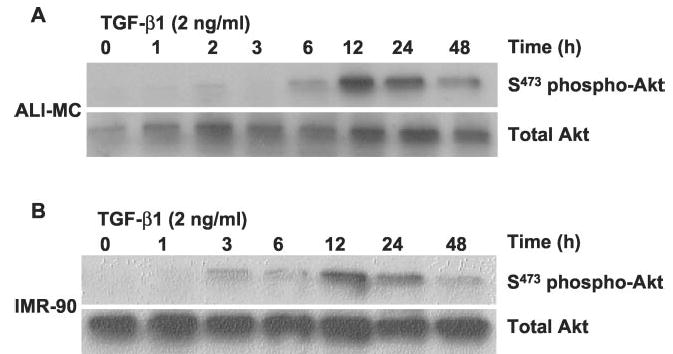
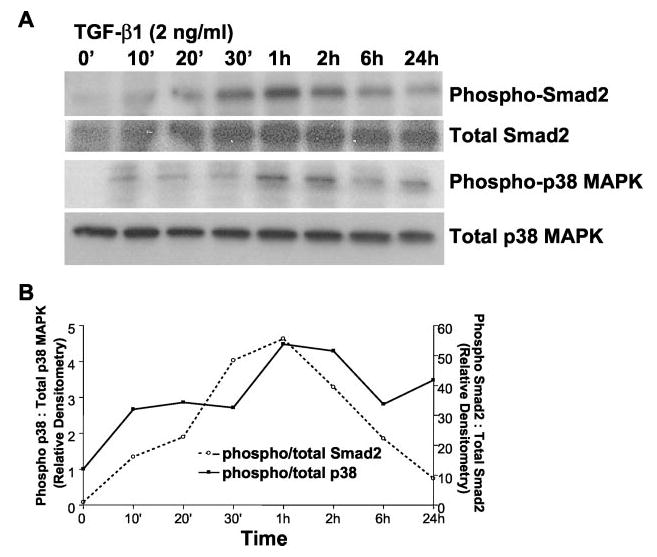
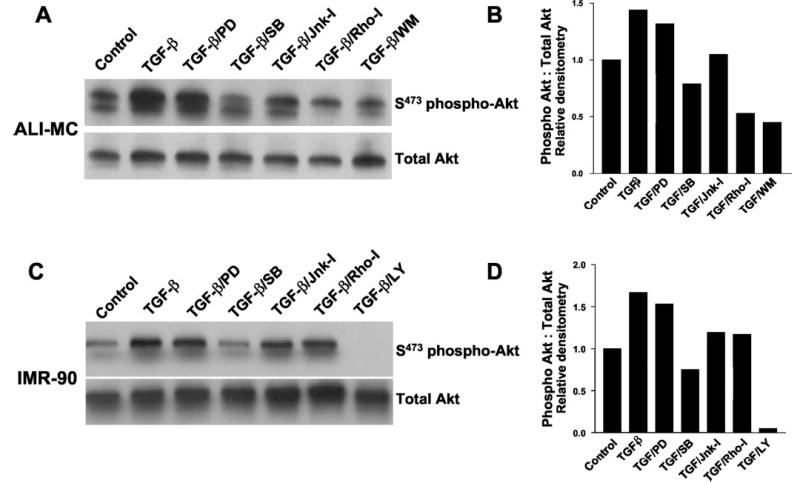
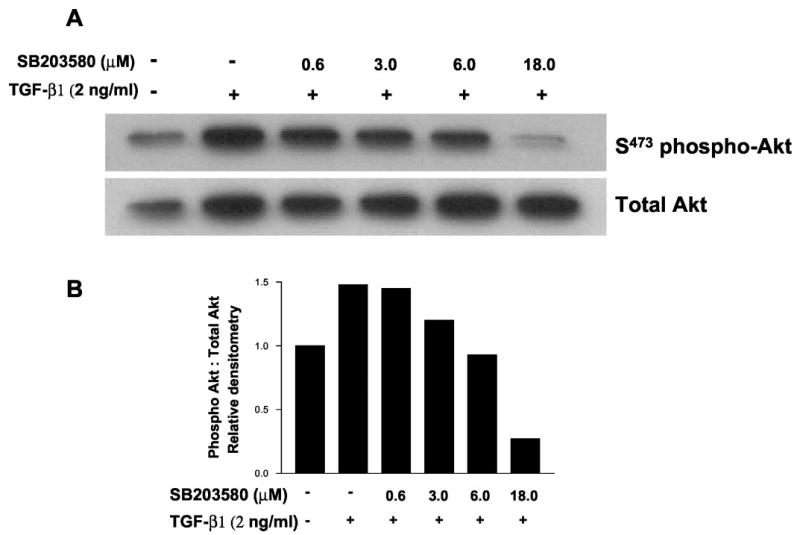
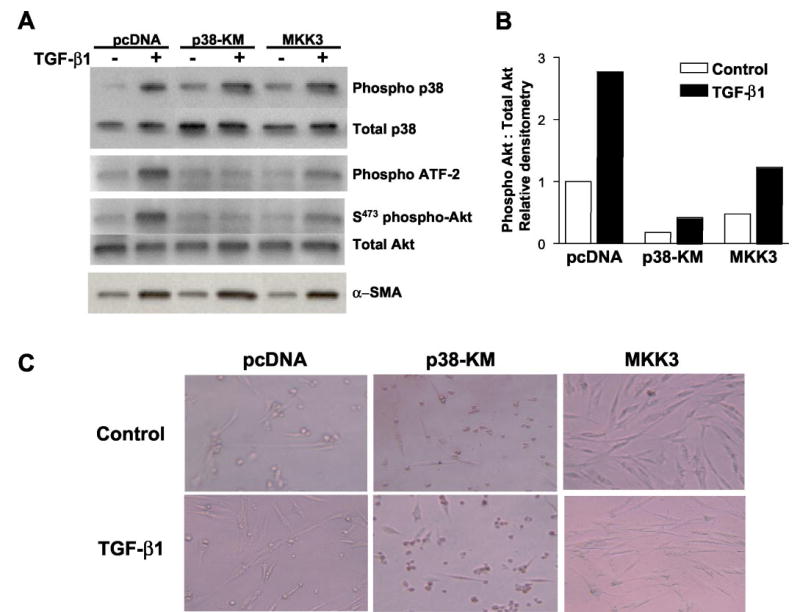
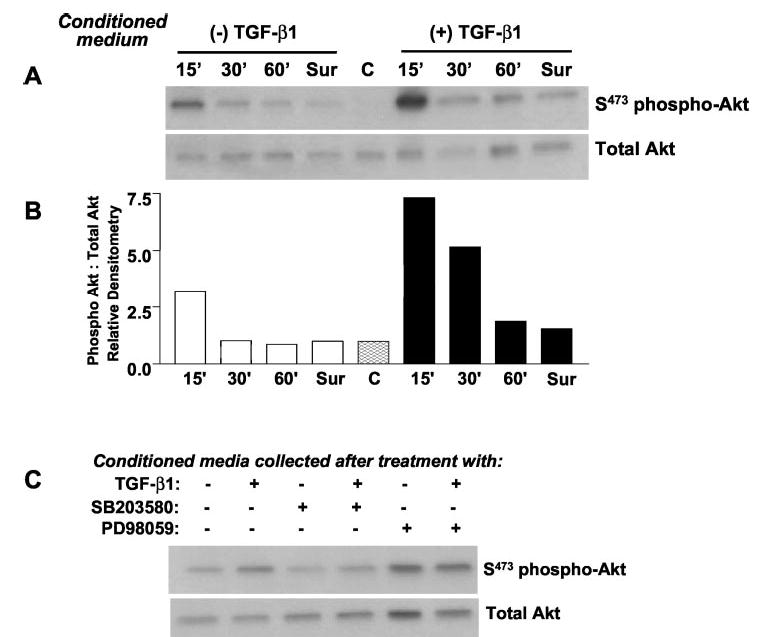
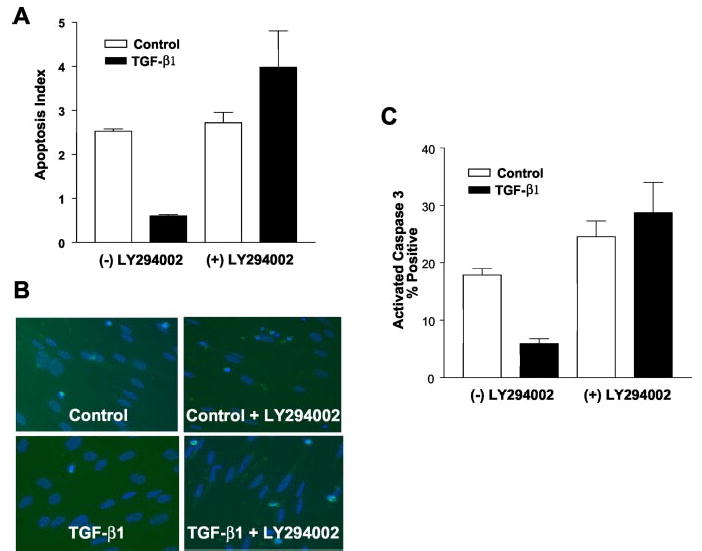
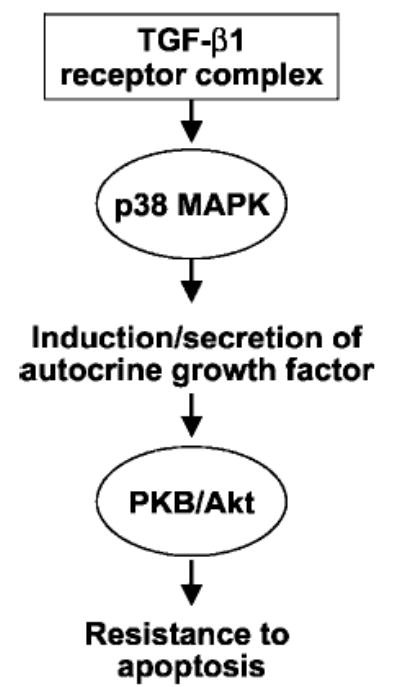
Transforming growth factor-beta1 (TGF-beta1) is a multifunctional cytokine involved in differentiation, growth, and survival of mesenchymal cells while inhibiting growth/survival of most other cell types. The mechanism(s) of pro-survival signaling by TGF-beta1 in mesenchymal cells is unclear. In this report, we demonstrate that TGF-beta1 protects against serum deprivation-induced apoptosis of mesenchymal cells isolated from patients with acute lung injury and of normal human fetal lung fibroblasts (IMR-90). TGF-beta receptor(s)-activated signaling in these cells involves rapid activation of the Smad and p38 MAPK pathways within minutes of TGF-beta1 treatment followed by a more delayed activation of the pro-survival phosphatidylinositol 3-kinase-protein kinase B (PKB)/Akt pathway. Pharmacological inhibition of p38 MAPK with SB203580 or expression of a p38 kinase-deficient mutant protein inhibits TGF-beta1-induced PKB/Akt phosphorylation. Conditioned medium from TGF-beta1-treated cells rapidly induces PKB/Akt activation in an SB203580- and suramin-sensitive manner, suggesting p38 MAPK-dependent production of a secreted growth factor that activates this pro-survival pathway by an autocrine/paracrine mechanism. Inhibition of the phosphatidylinositol 3-kinase-PKB/Akt pathway blocks TGF-beta1-induced resistance to apoptosis. These results demonstrate the activation of a novel TGF-beta1-activated pro-survival/anti-apoptotic signaling pathway in mesenchymal cells/fibroblasts that may explain cell-specific actions of TGF-beta1 and provide mechanistic insights into its pro-fibrotic and tumor-promoting effects.
Figures
References
-
- Massague J. Nat Rev Mol Cell Biol. 2000;1:169–178. - PubMed
-
- Roberts, A. B., and Derynck, R. (2001) Science’s STKE http:/www.stke.org/cgi/content/full/OC_sigtrans;2001/PE43 - PubMed
-
- Grande JP. Proc Soc Exp Biol Med. 1997;214:27–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
