

Unifying features in protein-folding mechanisms
- PMID: 14595026
- PMCID: PMC263785
- DOI: 10.1073/pnas.1835776100
Unifying features in protein-folding mechanisms
Abstract
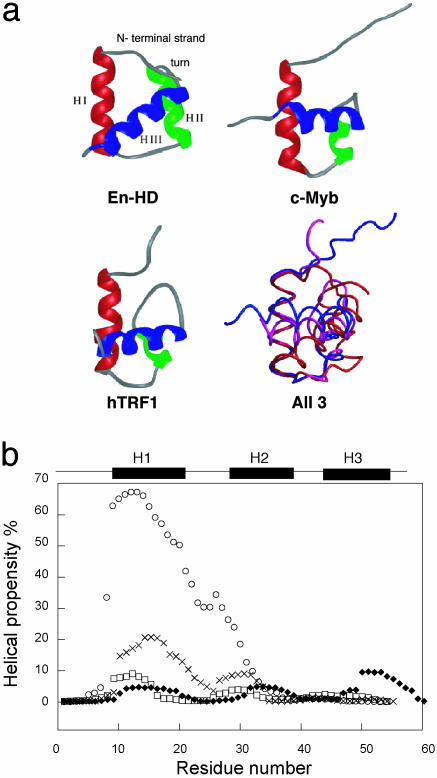
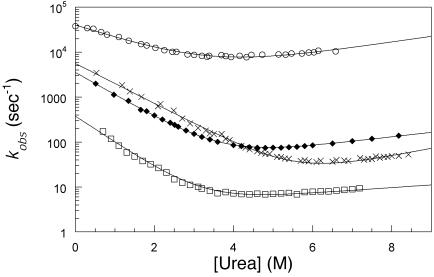
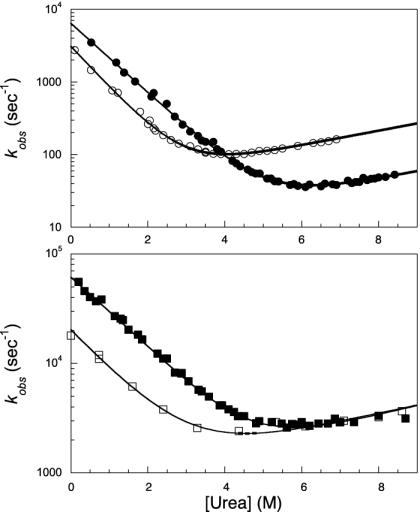
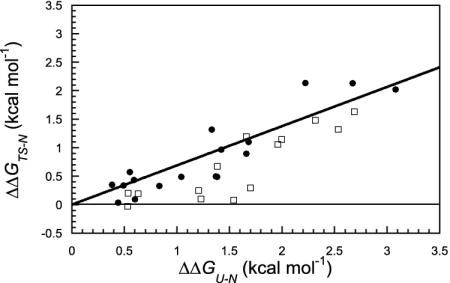
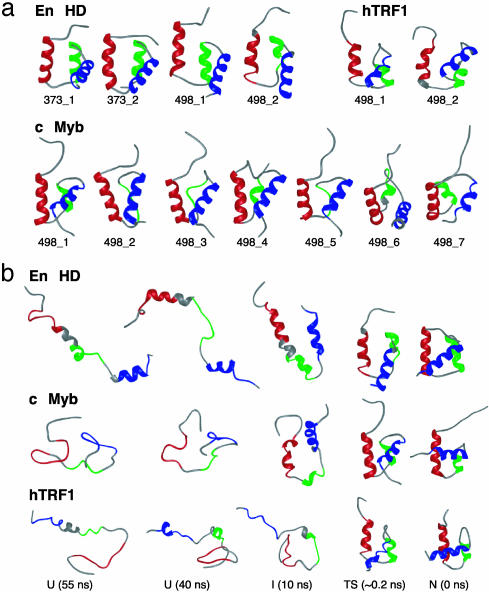
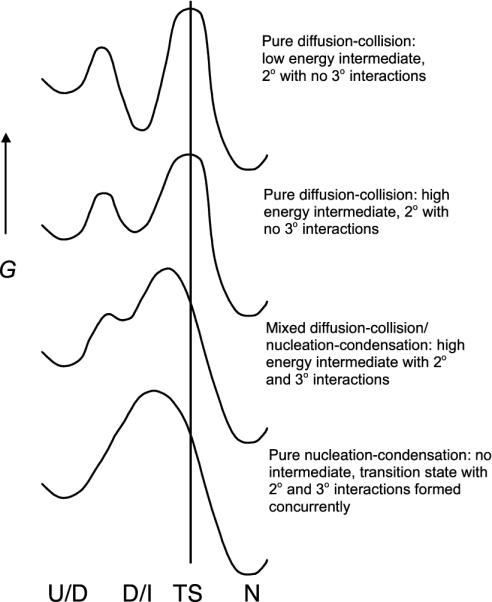
We compare the folding of representative members of a protein superfamily by experiment and simulation to investigate common features in folding mechanisms. The homeodomain superfamily of three-helical, single-domain proteins exhibits a spectrum of folding processes that spans the complete transition from concurrent secondary and tertiary structure formation (nucleation-condensation mechanism) to sequential secondary and tertiary formation (framework mechanism). The unifying factor in their mechanisms is that the transition state for (un)folding is expanded and very native-like, with the proportion and degree of formation of secondary and tertiary interactions varying. There is a transition, or slide, from the framework to nucleation-condensation mechanism with decreasing stability of the secondary structure. Thus, framework and nucleation-condensation are different manifestations of an underlying common mechanism.
Figures
References
-
- Myers, J. K. & Oas, T. G. (1999) Biochemistry 38, 6761-6768. - PubMed
-
- Mayor, U., Guydosh N. R., Johnson C. M., Grossmann J. G., Sato S., Jas G. S., Freund S. M., Alonso D. O., Daggett V. & Fersht, A. R. (2003) Nature 421, 863-867. - PubMed
-
- Islam, S. A., Karplus, M. & Weaver, D. L. (2002) J. Mol. Biol. 318, 199-215. - PubMed
-
- Kim, P. & Baldwin, R. L. (1990) Annu. Rev. Biochem. 59, 631-660. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
