

Mitochondrial DNA mutations in human colonic crypt stem cells
- PMID: 14597761
- PMCID: PMC228466
- DOI: 10.1172/JCI19435
Mitochondrial DNA mutations in human colonic crypt stem cells
Abstract
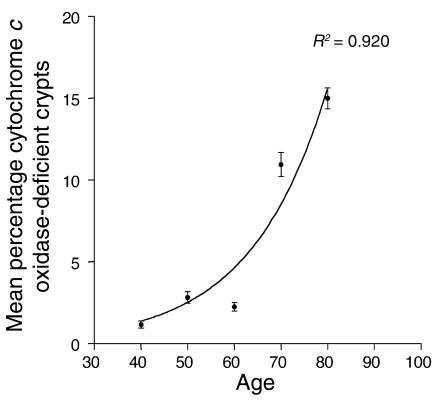
The mitochondrial genome encodes 13 essential subunits of the respiratory chain and has remarkable genetics based on uniparental inheritance. Within human populations, the mitochondrial genome has a high rate of sequence divergence with multiple polymorphic variants and thus has played a major role in examining the evolutionary history of our species. In recent years it has also become apparent that pathogenic mitochondrial DNA (mtDNA) mutations play an important role in neurological and other diseases. Patients harbor many different mtDNA mutations, some of which are mtDNA mutations, some of which are inherited, but others that seem to be sporadic. It has also been suggested that mtDNA mutations play a role in aging and cancer, but the evidence for a causative role in these conditions is less clear. The accumulated data would suggest, however, that mtDNA mutations occur on a frequent basis. In this article we describe a new phenomenon: the accumulation of mtDNA mutations in human colonic crypt stem cells that result in a significant biochemical defect in their progeny. These studies have important consequences not only for understanding of the finding of mtDNA mutations in aging tissues and tumors, but also for determining the frequency of mtDNA mutations within a cell.
Figures
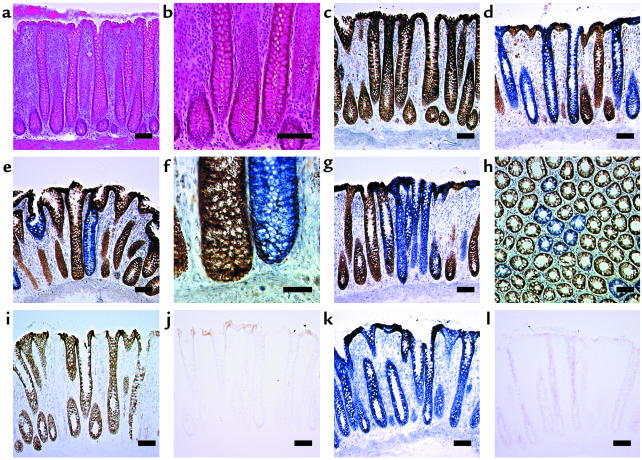
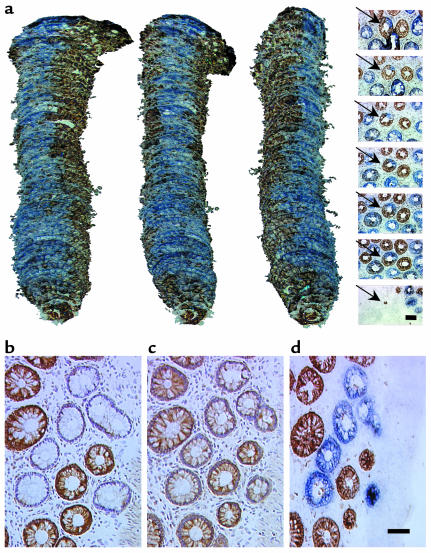
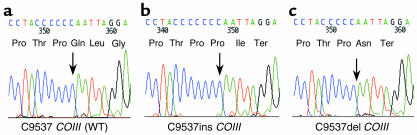
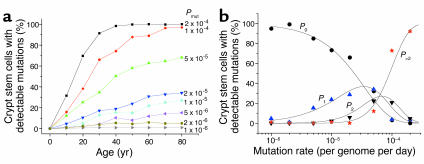
Comment in
-
Tales from the crypt.J Clin Invest. 2003 Nov;112(9):1312-6. doi: 10.1172/JCI20249. J Clin Invest. 2003. PMID: 14597757 Free PMC article.
References
-
- Chinnery PF, et al. The epidemiology of pathogenic mitochondrial DNA mutations. Ann. Neurol. 2000;48:188–193. - PubMed
-
- Lightowlers RN, Chinnery PF, Turnbull DM, Howell N. Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet. 1997;13:450–455. - PubMed
-
- Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum. Mol. Genet. 1994;3:13–19. - PubMed
-
- Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann. Neurol. 1998;43:217–223. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
