

NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis
- PMID: 14597764
- PMCID: PMC228420
- DOI: 10.1172/JCI18212
NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis
Abstract
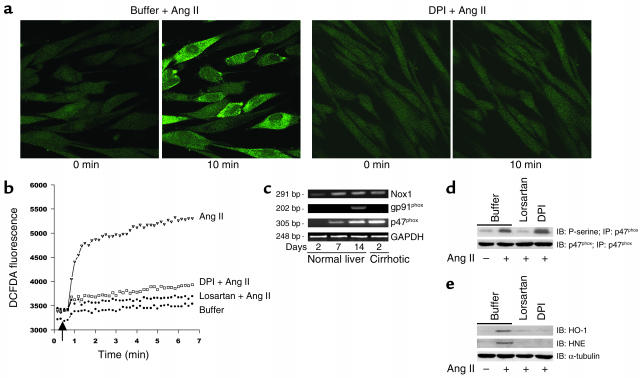
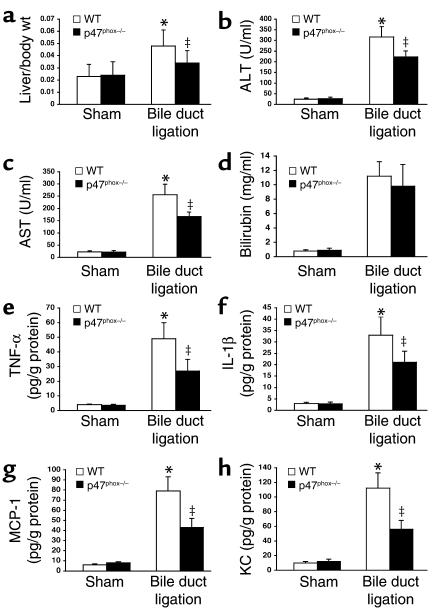
Angiotensin II (Ang II) is a pro-oxidant and fibrogenic cytokine. We investigated the role of NADPH oxidase in Ang II-induced effects in hepatic stellate cells (HSCs), a fibrogenic cell type. Human HSCs express mRNAs of key components of nonphagocytic NADPH oxidase. Ang II phosphorylated p47phox, a regulatory subunit of NADPH oxidase, and induced reactive oxygen species formation via NADPH oxidase activity. Ang II phosphorylated AKT and MAPKs and increased AP-1 DNA binding in a redox-sensitive manner. Ang II stimulated DNA synthesis, cell migration, procollagen alpha1(I) mRNA expression, and secretion of TGF-beta1 and inflammatory cytokines. These effects were attenuated by N-acetylcysteine and diphenylene iodonium, an NADPH oxidase inhibitor. Moreover, Ang II induced upregulation of genes potentially involved in hepatic wound-healing response in a redox-sensitive manner, as assessed by microarray analysis. HSCs isolated from p47phox-/- mice displayed a blunted response to Ang II compared with WT cells. We also assessed the role of NADPH oxidase in experimental liver fibrosis. After bile duct ligation, p47phox-/- mice showed attenuated liver injury and fibrosis compared with WT counterparts. Moreover, expression of smooth muscle alpha-actin and expression of TGF-beta1 were reduced in p47phox-/- mice. Thus, NADPH oxidase mediates the actions of Ang II on HSCs and plays a critical role in liver fibrogenesis.
Figures





Comment in
-
"HOPE" for the liver? Mechanistic insight into the role of the renin-angiotensin system in hepatic fibrosis.Hepatology. 2004 Jul;40(1):263-5. doi: 10.1002/hep.20296. Hepatology. 2004. PMID: 15239111 No abstract available.
References
-
- Albanis E, Friedman SL. Hepatic fibrosis. Pathogenesis and principles of therapy. Clin. Liver Dis. 2001;5:315–334. - PubMed
-
- Bataller R, Brenner DA. Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin. Liver Dis. 2001;21:437–451. - PubMed
-
- Asbert M, et al. Assessment of the renin-angiotensin system in cirrhotic patients. Comparison between plasma renin activity and direct measurement of immunoreactive renin. J. Hepatol. 1992;15:179–183. - PubMed
-
- Paizis G, et al. Up-regulation of components of the renin-angiotensin system in the bile duct-ligated rat liver. Gastroenterology. 2002;123:1667–1676. - PubMed
-
- Powell EE, et al. Host genetic factors influence disease progression in chronic hepatitis C. Hepatology. 2000;31:828–833. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
