

Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells
- PMID: 14610056
- PMCID: PMC2173638
- DOI: 10.1083/jcb.200306001
Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells
Abstract
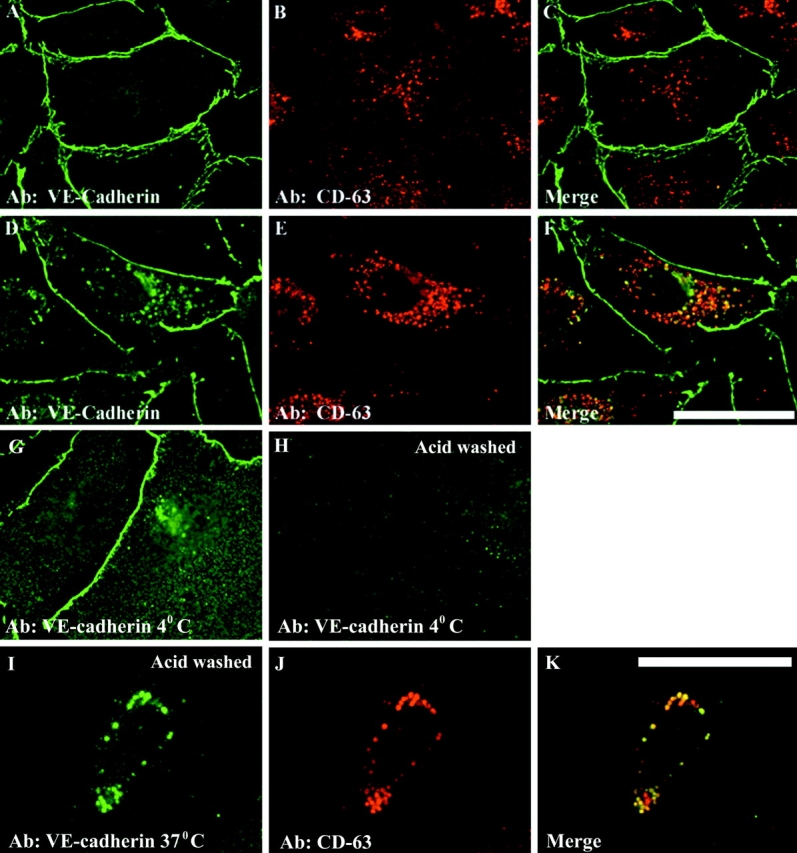
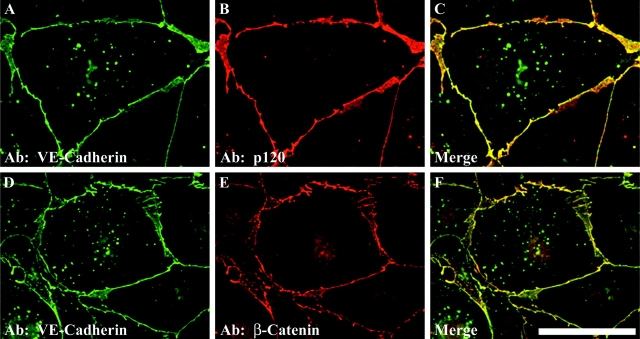
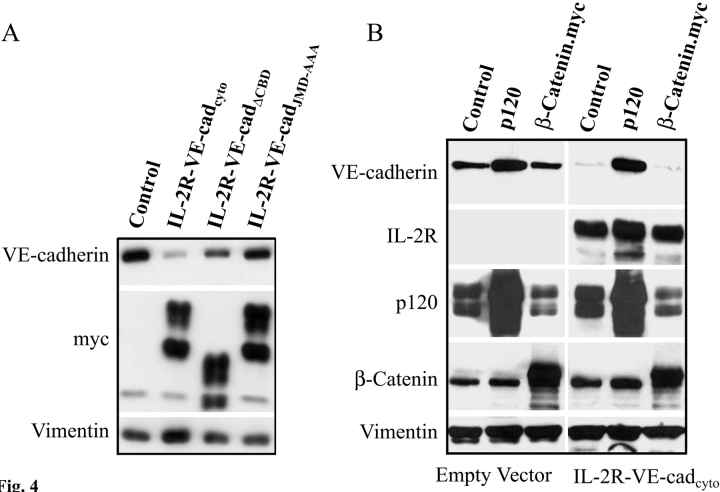
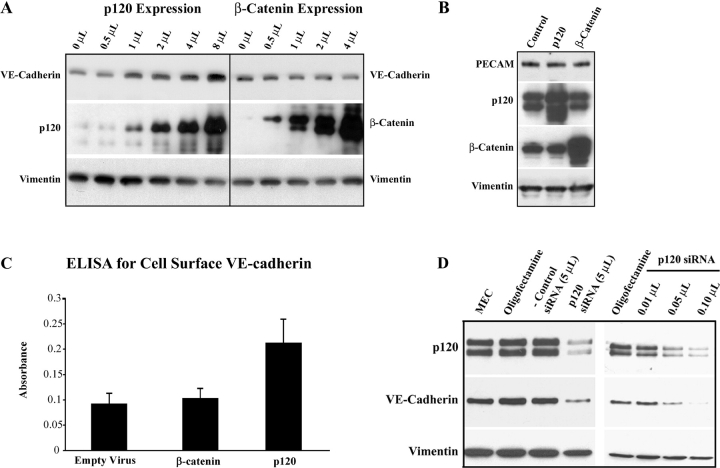
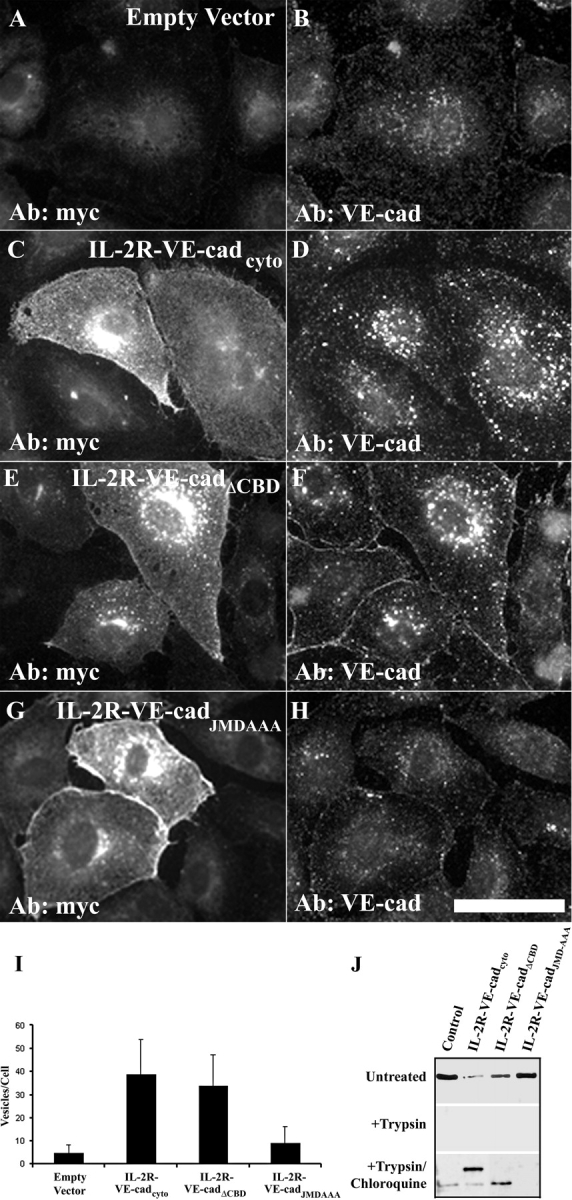
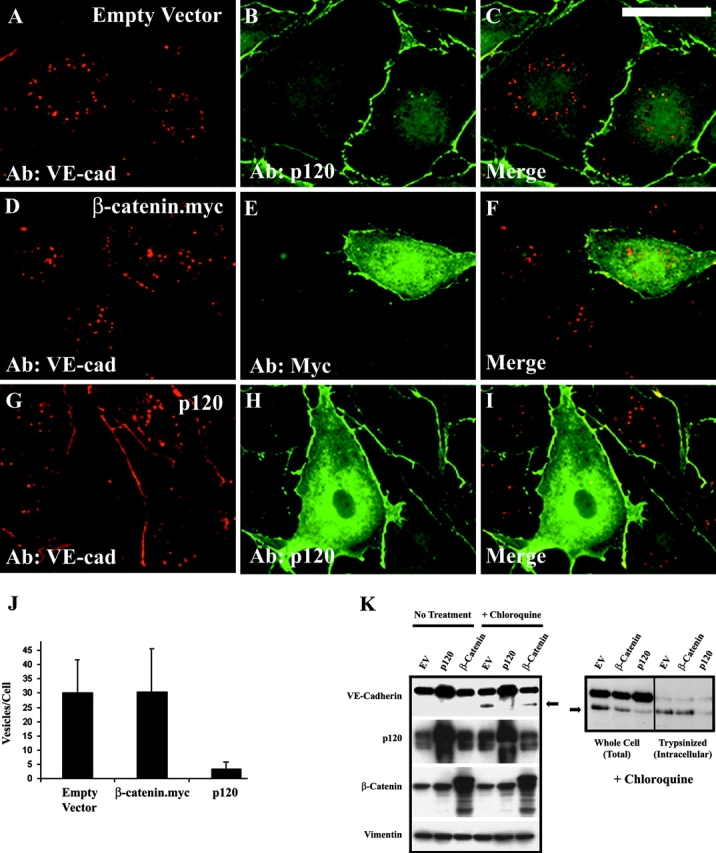
The mechanisms by which catenins regulate cadherin function are not fully understood, and the precise function of p120 catenin (p120ctn) has remained particularly elusive. In microvascular endothelial cells, p120ctn colocalized extensively with cell surface VE-cadherin, but failed to colocalize with VE-cadherin that had entered intracellular degradative compartments. To test the possibility that p120ctn binding to VE-cadherin regulates VE-cadherin internalization, a series of approaches were undertaken to manipulate p120ctn availability to endogenous VE-cadherin. Expression of VE-cadherin mutants that competed for p120ctn binding triggered the degradation of endogenous VE-cadherin. Similarly, reducing levels of p120ctn using siRNA caused a dramatic and dose-related reduction in cellular levels of VE-cadherin. In contrast, overexpression of p120ctn increased VE-cadherin cell surface levels and inhibited entry of cell surface VE-cadherin into degradative compartments. These results demonstrate that cellular levels of p120ctn function as a set point mechanism that regulates cadherin expression levels, and that a major function of p120ctn is to control cadherin internalization and degradation.
Figures

Comment in
-
Traffic control: p120-catenin acts as a gatekeeper to control the fate of classical cadherins in mammalian cells.J Cell Biol. 2003 Nov 10;163(3):437-40. doi: 10.1083/jcb.200310090. J Cell Biol. 2003. PMID: 14610049 Free PMC article. Review.
References
-
- Aberle, H., S. Butz, J. Stappert, H. Weissig, R. Kemler, and H. Hoschuetzky. 1994. Assembly of the cadherin-catenin complex in vitro with recombinant proteins. J. Cell Sci. 107:3655–3663. - PubMed
-
- Anastasiadis, P.Z., and A.B. Reynolds. 2000. The p120 catenin family: complex roles in adhesion, signaling and cancer. J. Cell Sci. 113:1319–1334. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
