

Nonhomologous-end-joining factors regulate DNA repair fidelity during Sleeping Beauty element transposition in mammalian cells
- PMID: 14612396
- PMCID: PMC262663
- DOI: 10.1128/MCB.23.23.8505-8518.2003
Nonhomologous-end-joining factors regulate DNA repair fidelity during Sleeping Beauty element transposition in mammalian cells
Abstract
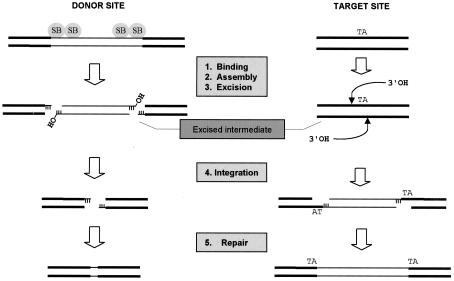
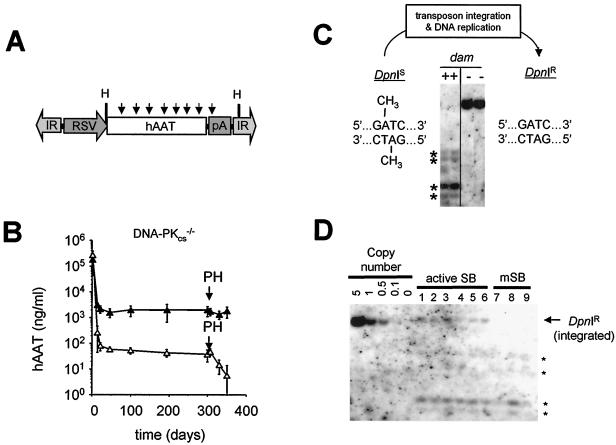
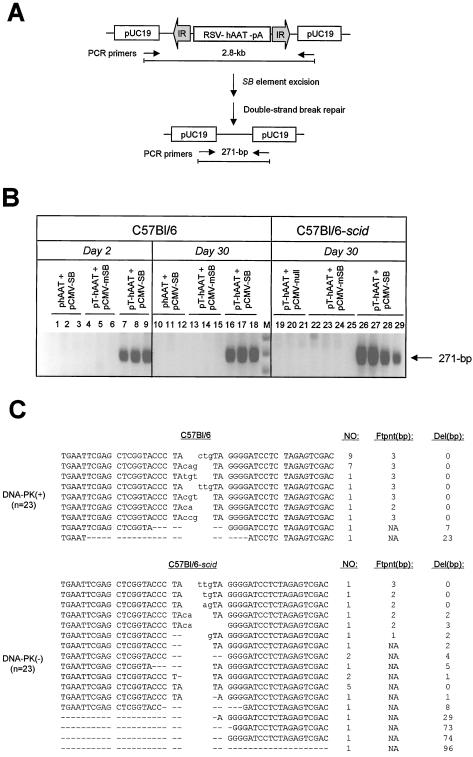
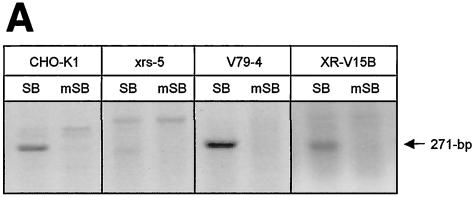
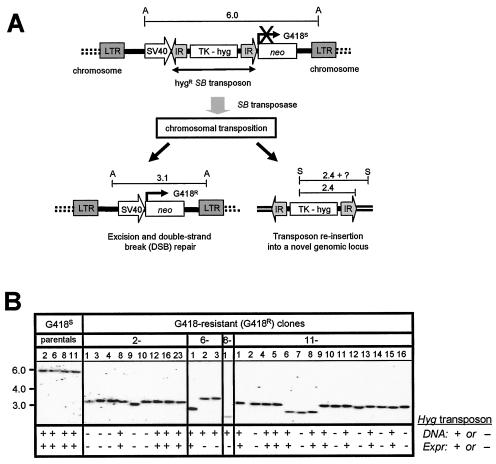
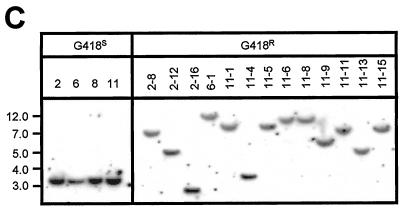
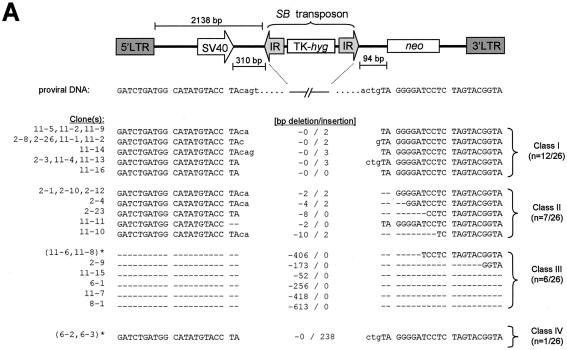
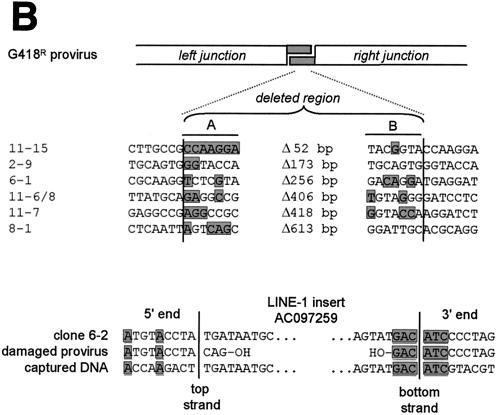
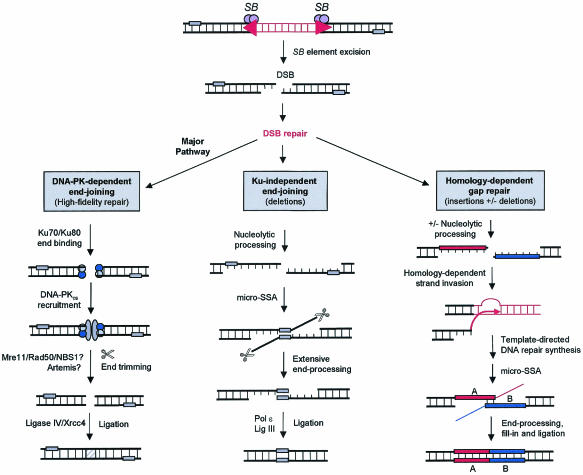
Herein, we report that the DNA-dependent protein kinase (DNA-PK) regulates the DNA damage introduced during Sleeping Beauty (SB) element excision and reinsertion in mammalian cells. Using both plasmid- and chromosome-based mobility assays, we analyzed the repair of transposase-induced double-stranded DNA breaks in cells deficient in either the DNA-binding subunit of DNA-PK (Ku) or its catalytic subunit (DNA-PKcs). We found that the free 3' overhangs left after SB element excision were efficiently and accurately processed by the major Ku-dependent nonhomologous-end-joining pathway. Rejoining of broken DNA molecules in the absence of Ku resulted in extensive end degradation at the donor site and greatly increased the frequency of recombination with ectopic templates. Therefore, the major DNA-PK-dependent DNA damage response predominates over more-error-prone repair pathways and thereby facilitates high-fidelity DNA repair during transposon mobilization in mammalian cells. Although transposable elements were not found to be efficiently circularized after transposase-mediated excision, DNA-PK deficiency supported more-frequent transposase-mediated element insertion than was found in wild-type controls. We conclude that, based on its ability to regulate excision site junctional diversity and transposon insertion frequency, DNA-PK serves an important protective role during transpositional recombination in mammals.
Figures

References
-
- Adams, M. D., M. McVey, and J. J. Sekelsky. 2003. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science 299:265-267. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous