

Platelet and osteoclast beta3 integrins are critical for bone metastasis
- PMID: 14612570
- PMCID: PMC283570
- DOI: 10.1073/pnas.2234372100
Platelet and osteoclast beta3 integrins are critical for bone metastasis
Abstract
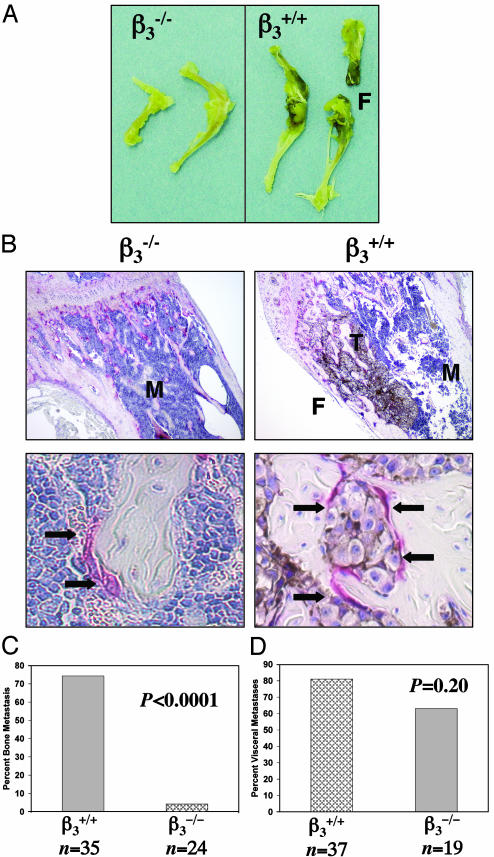
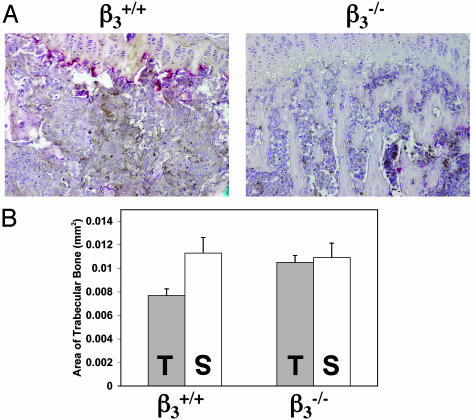
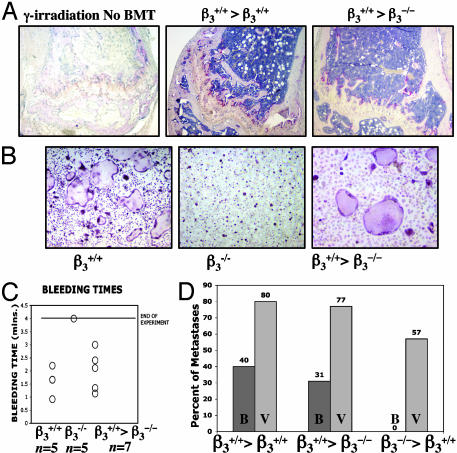
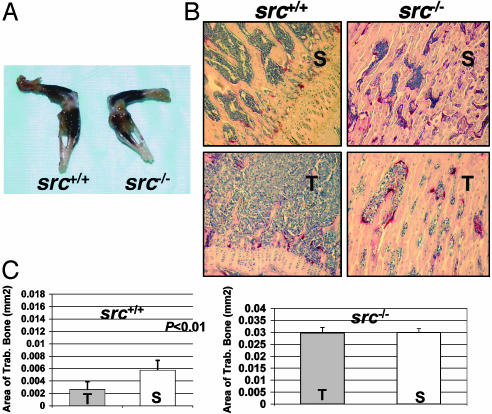
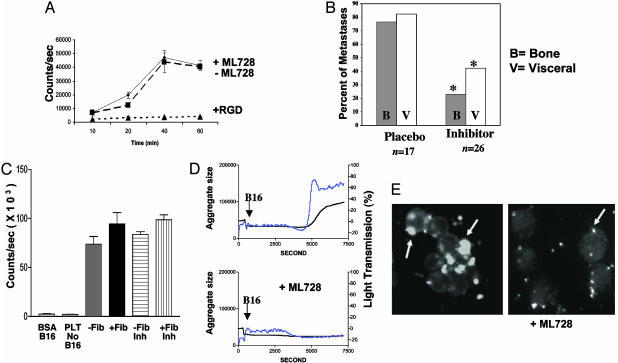
Mice with a targeted deletion of beta3 integrin were used to examine the process by which tumor cells metastasize and destroy bone. Injection of B16 melanoma cells into the left cardiac ventricle resulted in osteolytic bone metastasis in 74% of beta3+/+ mice by 14 days. In contrast, only 4% of beta3-/- mice developed bone lesions. Direct intratibial inoculation of tumor resulted in marrow replacement by tumor in beta3-/- mice, but no associated trabecular bone resorption as seen inbeta3+/+ mice. Bone marrow transplantation studies showed that susceptibility to bone metastasis was conferred by a bone marrow-derived cell. To dissect the roles of osteoclast and platelet beta3 integrins in this model of bone metastasis, osteoclast-defective src-/- mice were used. Src-null mice were protected from tumor-associated bone destruction but were not protected from tumor cell metastasis to bone. In contrast, a highly specific platelet aggregation inhibitor of activated alphaIIbbeta3 prevented B16 metastases. These data demonstrate a critical role for platelet alphaIIbbeta3 in tumor entry into bone and suggest a mechanism by which antiplatelet therapy may be beneficial in preventing the metastasis of solid tumors.
Figures
Similar articles
-
Critical role of beta3 integrin in experimental postmenopausal osteoporosis.J Bone Miner Res. 2005 Dec;20(12):2116-23. doi: 10.1359/JBMR.050724. Epub 2005 Jul 25. J Bone Miner Res. 2005. PMID: 16294265
-
CD47 regulates bone mass and tumor metastasis to bone.Cancer Res. 2009 Apr 1;69(7):3196-204. doi: 10.1158/0008-5472.CAN-08-3358. Epub 2009 Mar 10. Cancer Res. 2009. PMID: 19276363 Free PMC article.
-
Dissection of platelet and myeloid cell defects by conditional targeting of the beta3-integrin subunit.FASEB J. 2010 Apr;24(4):1117-27. doi: 10.1096/fj.09-138420. Epub 2009 Nov 20. FASEB J. 2010. PMID: 19933310 Free PMC article.
-
Integrins and bone metastasis: integrating tumor cell and stromal cell interactions.Bone. 2011 Jan;48(1):54-65. doi: 10.1016/j.bone.2010.09.016. Epub 2010 Sep 17. Bone. 2011. PMID: 20850578 Free PMC article. Review.
-
Basic mechanisms responsible for osteolytic and osteoblastic bone metastases.Clin Cancer Res. 2006 Oct 15;12(20 Pt 2):6213s-6216s. doi: 10.1158/1078-0432.CCR-06-1007. Clin Cancer Res. 2006. PMID: 17062703 Review.
Cited by
-
Antagonizing Integrin β3 Increases Immunosuppression in Cancer.Cancer Res. 2016 Jun 15;76(12):3484-95. doi: 10.1158/0008-5472.CAN-15-2663. Epub 2016 May 23. Cancer Res. 2016. PMID: 27216180 Free PMC article.
-
miRNA-34c Suppresses Osteosarcoma Progression In Vivo by Targeting Notch and E2F.JBMR Plus. 2022 Apr 9;6(5):e10623. doi: 10.1002/jbm4.10623. eCollection 2022 May. JBMR Plus. 2022. PMID: 35509638 Free PMC article.
-
Integrins in cancer: biological implications and therapeutic opportunities.Nat Rev Cancer. 2010 Jan;10(1):9-22. doi: 10.1038/nrc2748. Nat Rev Cancer. 2010. PMID: 20029421 Free PMC article. Review.
-
Contribution of platelets to tumour metastasis.Nat Rev Cancer. 2011 Feb;11(2):123-34. doi: 10.1038/nrc3004. Nat Rev Cancer. 2011. PMID: 21258396 Free PMC article. Review.
-
The Platelet Lifeline to Cancer: Challenges and Opportunities.Cancer Cell. 2018 Jun 11;33(6):965-983. doi: 10.1016/j.ccell.2018.03.002. Epub 2018 Apr 12. Cancer Cell. 2018. PMID: 29657130 Free PMC article. Review.
References
-
- Mundy, G. R. (2002) Nat. Rev. Cancer 2, 584–593. - PubMed
-
- Clohisy, D. R. & Ramnaraine, M. L. (1998) J. Orthop. Res. 16, 660–666. - PubMed
-
- Lipton, A., Theriault, R. L., Hortobagyi, G. N., Simeone, J., Knight, R. D., Mellars, K., Reitsma, D. J., Heffernan, M. & Seaman, J. J. (2000) Cancer 88, 1082–1090. - PubMed
-
- Yoneda, T., Michigami, T., Yi, B., Williams, P. J., Niewolna, M. & Hiraga, T. (2000) Cancer 88, 2979–2988. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
