

Pathogenesis of X-linked Charcot-Marie-Tooth disease: differential effects of two mutations in connexin 32
- PMID: 14627639
- PMCID: PMC4513672
- DOI: 10.1523/JNEUROSCI.23-33-10548.2003
Pathogenesis of X-linked Charcot-Marie-Tooth disease: differential effects of two mutations in connexin 32
Abstract
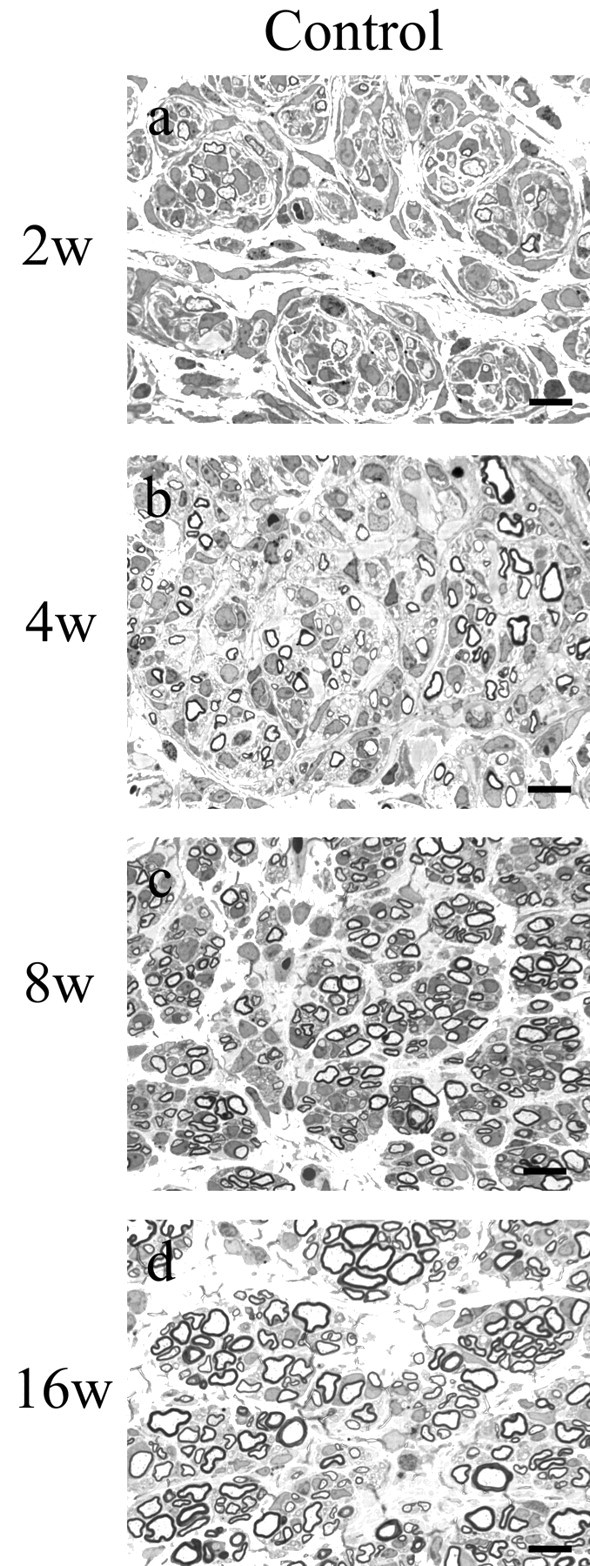
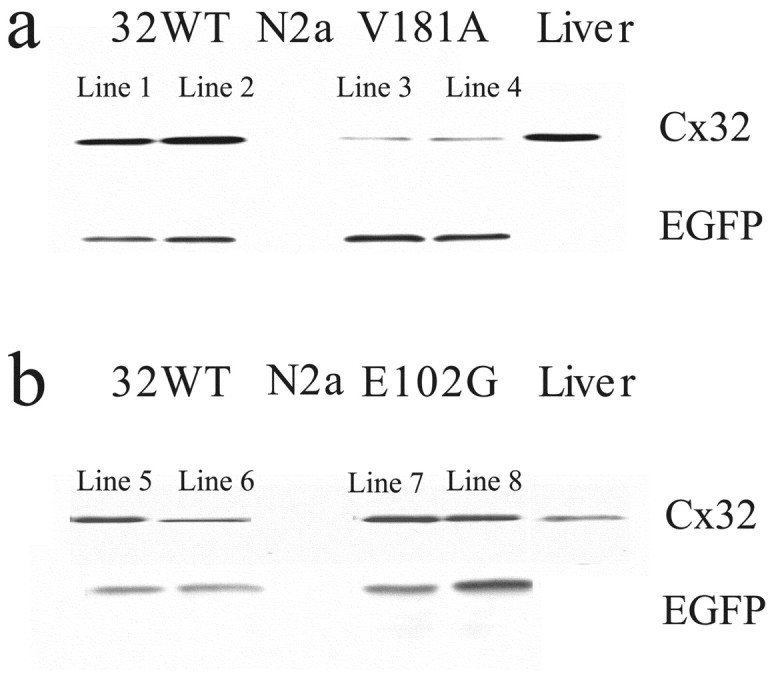
X-linked Charcot-Marie-Tooth disease is an inherited peripheral neuropathy arising in patients with mutations in the gene encoding connexin 32 (Cx32). Cx32 is expressed at the paranodes and Schmidt-Lantermann incisures of myelinating Schwann cells in which it is believed to form a reflexive pathway between the abaxonal and adaxonal cytoplasmic domains. Patients with the Val181Ala (V181A) mutation have a severe peripheral neuropathy. Experiments using a nude mouse xenograft system show that Schwann cells expressing only this mutant form of Cx32 are profoundly impaired in their ability to support the earliest stages of regeneration of myelinated fibers. Coupling between paired Xenopus oocytes expressing V181A is reduced compared with the coupling between oocytes expressing wild-type human Cx32 (32WT), and protein levels assayed by Western blot are substantially lower. Immunocytochemisty shows that Neuro2a cells expressing the V181A mutant have very few gap junction plaques compared with cells expressing 32WT; Cx32 protein levels are lower in these cells than in those expressing 32WT. Because failure of normal regeneration is evident before formation of myelin, loss of function of Cx32 may impact on the function of precursors of the myelinating Schwann cell before the formation of the hypothesized reflexive pathway. The Glu102Gly (E102G) mutation leads to a milder phenotype. Early regeneration is normal in grafts with Schwann cells expressing the E102G mutant. The only abnormality detected in the behavior of its channel is increased sensitivity to acidification-induced closure, a property that may lead to reduced gap junction coupling during periods of metabolic stress. This restricted functional abnormality may explain the relatively mild phenotype seen in the xenograft model and in E102G patients.
Figures









References
-
- Abrams CK, Bennett MVL ( 2000) Hereditary human diseases caused by connexin mutations. In: Gap junctions—molecular basis of cell communication in health and disease (Peracchia C, ed) pp 423-459. New York: Academic.
-
- Abrams CK, Freidin MM, Dobrenis K, Bargiello TA, Verselis VK, Bennett MVL, Sahenk Z ( 2000) Pathogenesis of CMTX: analysis of a new connexin 32 mutation leading to an inability to regenerate large caliber axons. Ann Neurol 48: 439.
-
- Abrams CK, Freidin MM, Verselis VK, Bennett MVL, Bargiello TA ( 2001) Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease. Brain Res 900: 9-25. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous