

Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair
- PMID: 14633615
- PMCID: PMC1892355
- DOI: 10.1016/S0002-9440(10)63598-5
Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair
Abstract
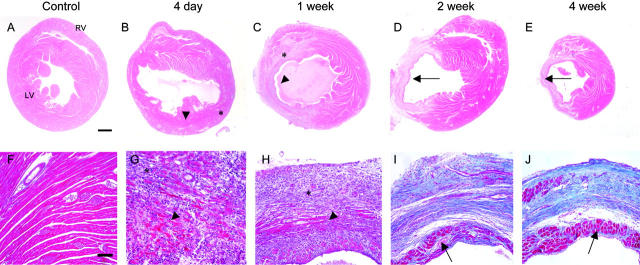
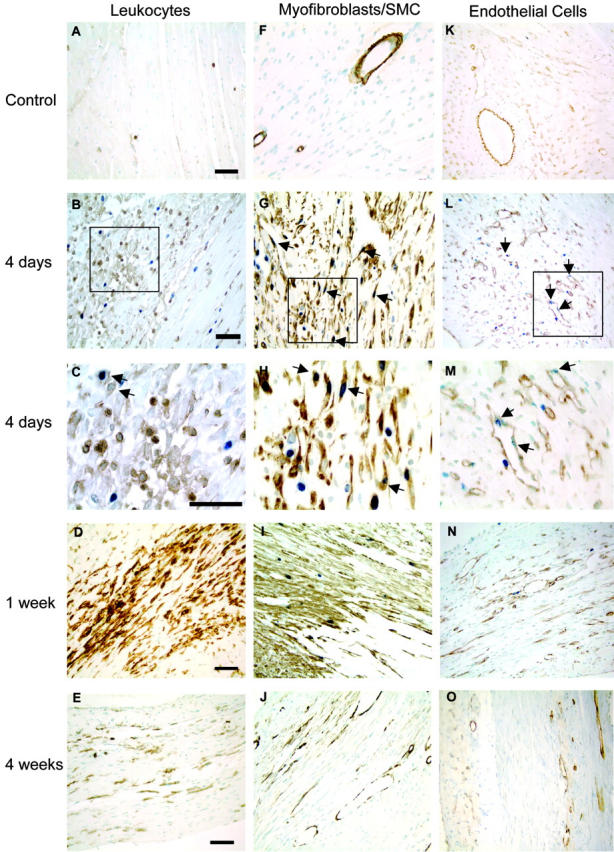
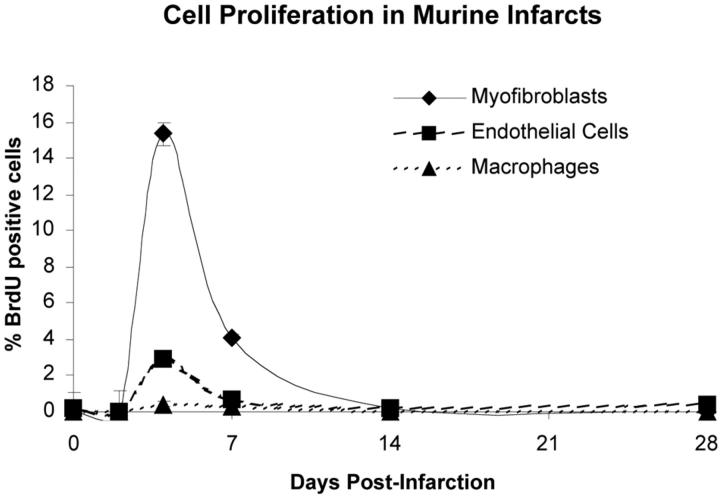
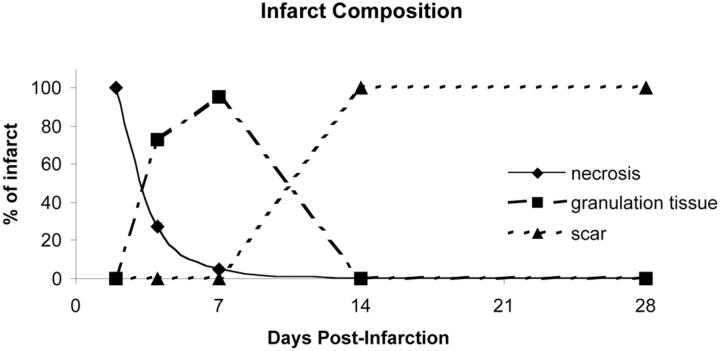
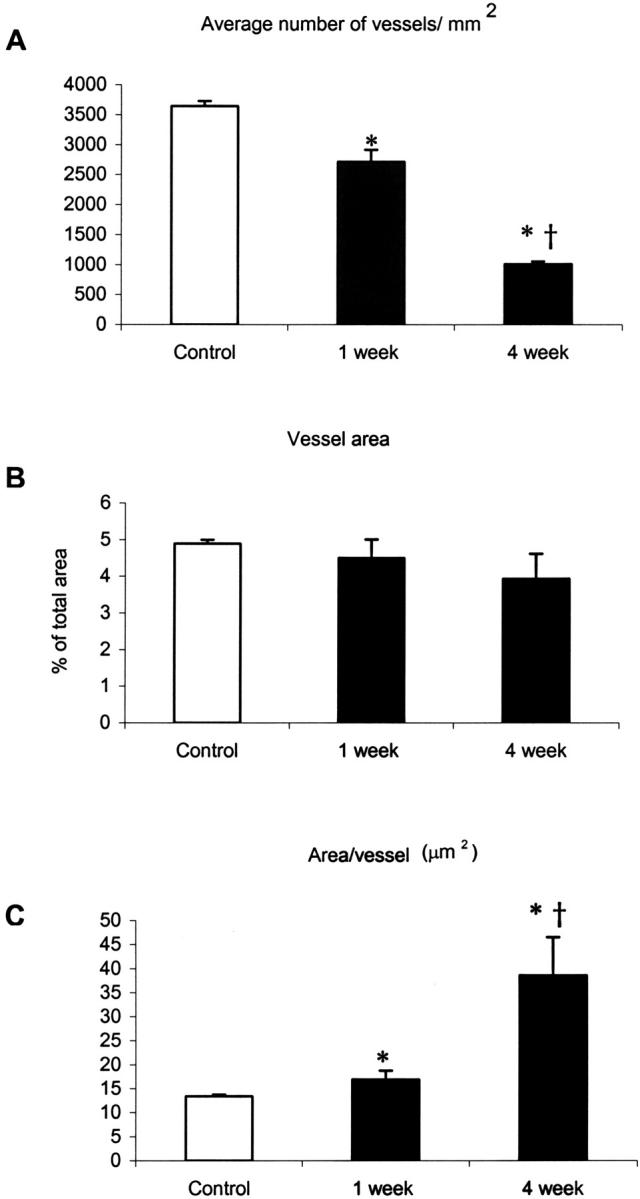
Granulation tissue formation is a critical step in infarct repair, however, the kinetics of cell replication and the molecules that regulate this process are poorly understood. In uninjured mouse hearts and at 2 days post-infarction, very little DNA synthesis (measured by incorporation of a BrdU pulse) was detected in any cell type. Four days after permanent coronary occlusion, the rates of myofibroblast (smooth muscle alpha-actin and BrdU double-positive) and endothelial cell (CD31 and BrdU double-positive) proliferation were 15.4 +/- 1.1% and 2.9 +/- 0.5%, respectively. Most proliferating cells were located at the interface of the infarct and viable tissue. By 1 week, fibroblast and endothelial cell proliferation declined to 4.1 +/- 0.6% and 0.7 +/- 0.1%, respectively. In the 2-week infarct, the remaining necrosis had been phagocytosed, and fibroblast and endothelial cell proliferation were <0.5%. Although leukocytes were abundant throughout infarct repair, no significant proliferation was detected at any time in cells expressing CD45 or mac-3. Infarct size at 4 days was 38 +/- 5% of the left ventricle and contracted to 20 +/- 4% by 4 weeks. After 4 days, the chamber dilated to four times that of the control hearts and remained so for the duration of the time course. The vascular density (per mm(2)) declined from 3643 +/- 82 in control hearts to 2716 +/- 197 at 1 week and 1010 +/- 47 at 4 weeks post-myocardial infarction (MI). The average percent area occupied by vessels did not change significantly between the groups but the area/vessel ( micro m(2)) increased from 14.1 +/- 0.3 in control hearts to 16.9 +/- 1.9 at 1 week and 38.7 +/- 7.9 at 4 weeks post-MI. These data indicate that mitogens for fibroblasts and endothelial cells peak within 4 days of infarction in the mouse heart. This provides the basis for identifying the responsible molecules and developing strategies to alter wound repair and improve cardiac function.
Figures
References
-
- Frangogiannis NG, Smith CW, Entman ML: The inflammatory response in myocardial infarction. Cardiovasc Res 2002, 53:31-47 - PubMed
-
- Cleutjens JPM: The role of matrix metalloproteinases in heart disease. Cardiovasc Res 1996, 32:816-821 - PubMed
-
- Bing RJ: Myocardial ischemia and infarction: growth of ideas. Cardiovasc Res 2001, 51:13-20 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
