

Osteocyte control of bone formation via sclerostin, a novel BMP antagonist
- PMID: 14633986
- PMCID: PMC291840
- DOI: 10.1093/emboj/cdg599
Osteocyte control of bone formation via sclerostin, a novel BMP antagonist
Abstract
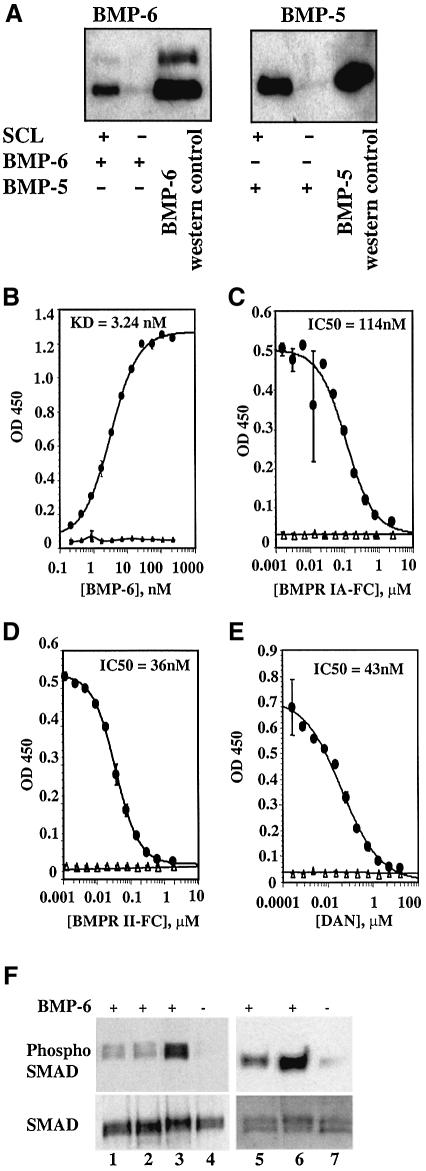
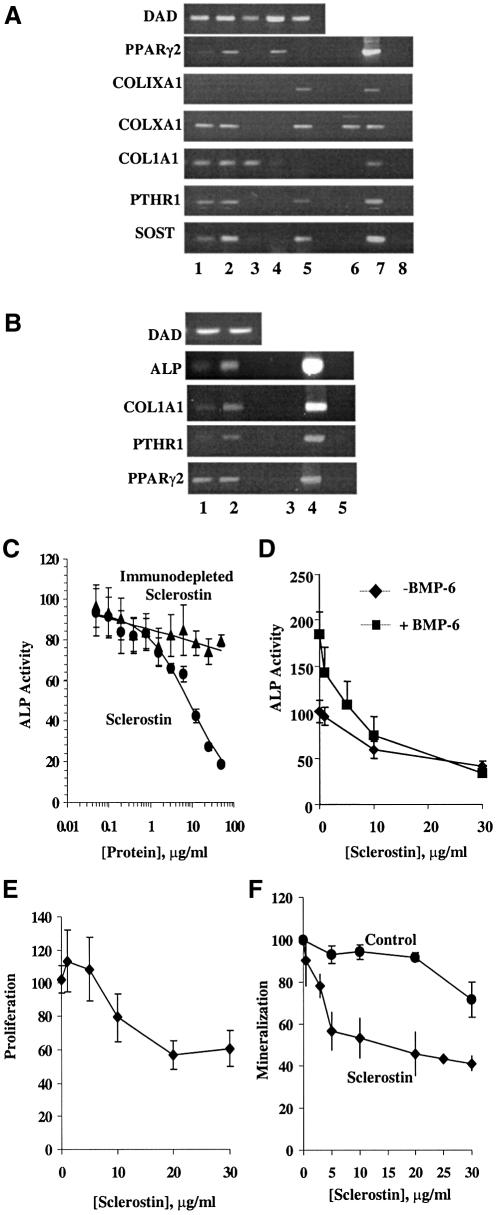
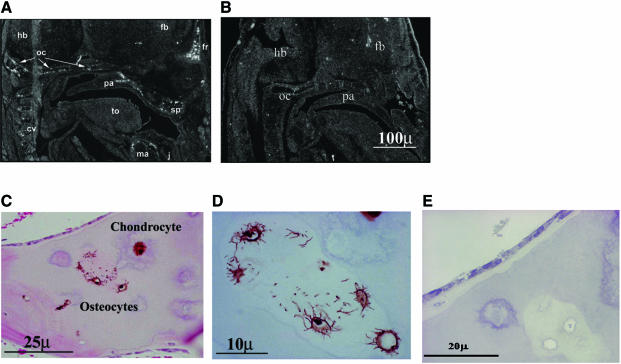
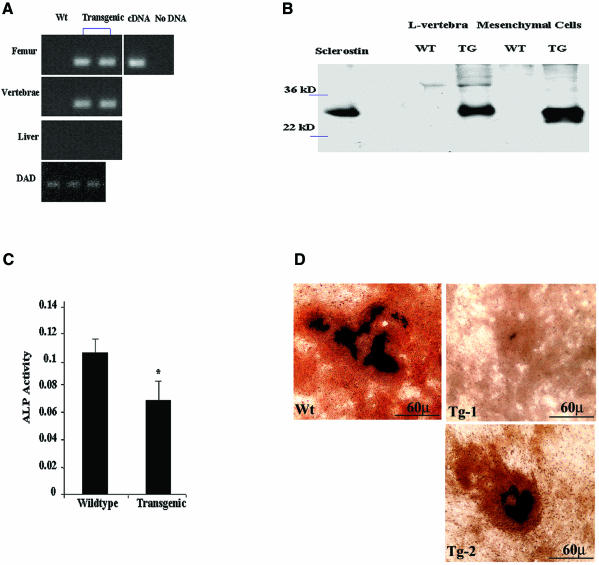
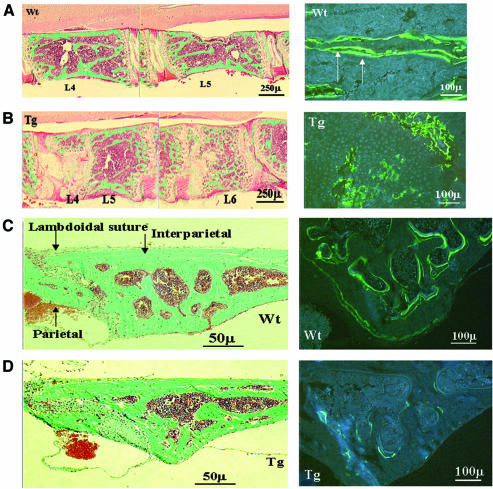
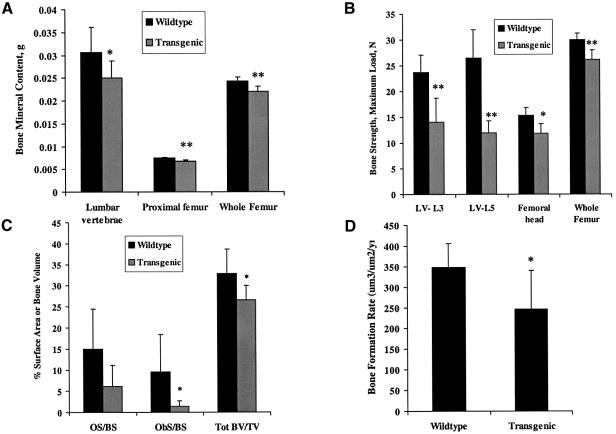
There is an unmet medical need for anabolic treatments to restore lost bone. Human genetic bone disorders provide insight into bone regulatory processes. Sclerosteosis is a disease typified by high bone mass due to the loss of SOST expression. Sclerostin, the SOST gene protein product, competed with the type I and type II bone morphogenetic protein (BMP) receptors for binding to BMPs, decreased BMP signaling and suppressed mineralization of osteoblastic cells. SOST expression was detected in cultured osteoblasts and in mineralizing areas of the skeleton, but not in osteoclasts. Strong expression in osteocytes suggested that sclerostin expressed by these central regulatory cells mediates bone homeostasis. Transgenic mice overexpressing SOST exhibited low bone mass and decreased bone strength as the result of a significant reduction in osteoblast activity and subsequently, bone formation. Modulation of this osteocyte-derived negative signal is therapeutically relevant for disorders associated with bone loss.
Figures
References
-
- Abe E., Yamamoto,M., Taguchi,Y., Lecka-Czernik,B., O’Brien,C.A., Economides,A.N., Stahl,N., Jilka,R.L. and Manolagas,S.C. (2000) Essential requirement of BMPs-2/4 for both osteoblast and osteoclast formation in murine bone marrow cultures from adult mice: antagonism by noggin. J. Bone Miner. Res., 15, 663–673. - PubMed
-
- Akhter M.P., Cullen,D.M., Gong,G. and Recker,R.R. (2001) Bone biomechanical properties in prostaglandin EP1 and EP2 knockout mice. Bone, 29, 121–125. - PubMed
-
- Balemans W. et al. (2001) Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet., 10, 537–543. - PubMed
-
- Balemans W., Foernzler,D., Parsons,C., Ebeling,M., Thompson,A., Reid,D.M., Lindpaintner,K., Ralston,S.H. and Van Hul,W. (2002) Lack of association between the SOST gene and bone mineral density in perimenopausal women: analysis of five polymorphisms. Bone, 31, 515–519. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
