

GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model
- PMID: 14640709
- PMCID: PMC2915448
- DOI: 10.1021/ac0347462
GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model
Abstract
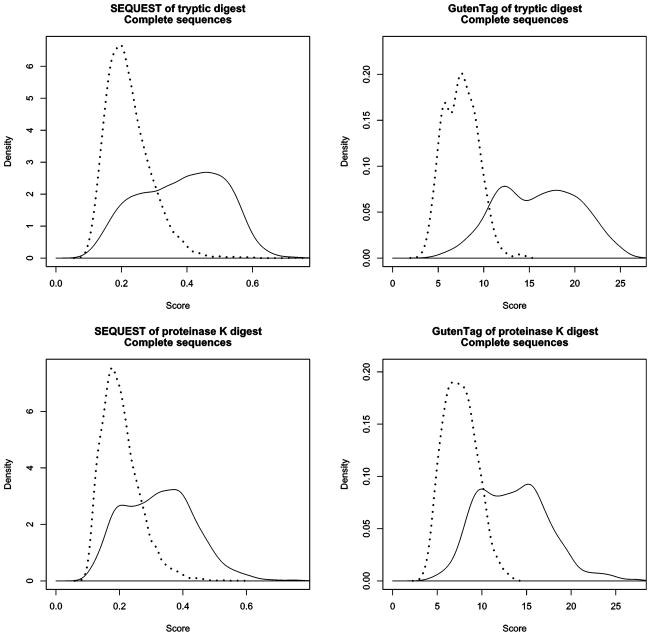
Shotgun proteomics is a powerful tool for identifying the protein content of complex mixtures via liquid chromatography and tandem mass spectrometry. The most widely used class of algorithms for analyzing mass spectra of peptides has been database search software such as SEQUEST. A new sequence tag database search algorithm, called GutenTag, makes it possible to identify peptides with unknown posttranslational modifications or sequence variations. This software automates the process of inferring partial sequence "tags" directly from the spectrum and efficiently examines a sequence database for peptides that match these tags. When multiple candidate sequences result from the database search, the software evaluates which is the best match by a rapid examination of spectral fragment ions. We compare GutenTag's accuracy to that of SEQUEST on a defined protein mixture, showing that both modified and unmodified peptides can be successfully identified by this approach. GutenTag analyzed 33,000 spectra from a human lens sample, identifying peptides that were missed in prior SEQUEST analysis due to sequence polymorphisms and posttranslational modifications. The software is available under license; visit http://fields.scripps.edu for information.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
