

Gene structure prediction in syntenic DNA segments
- PMID: 14654703
- PMCID: PMC291857
- DOI: 10.1093/nar/gkg905
Gene structure prediction in syntenic DNA segments
Abstract
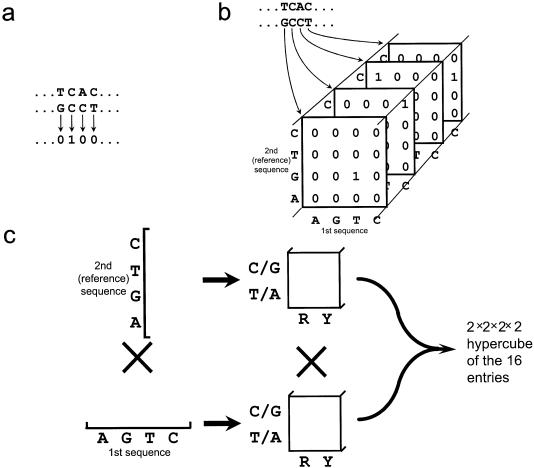
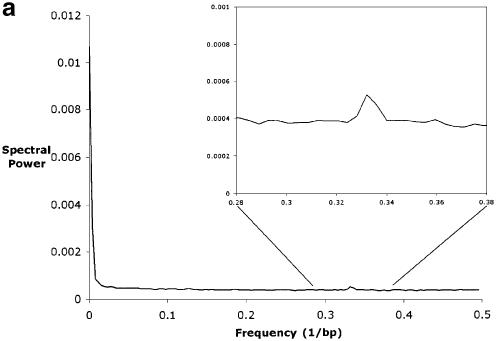
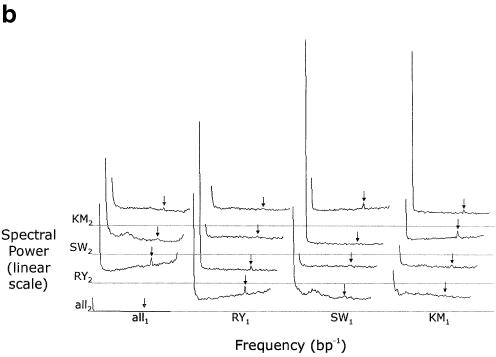
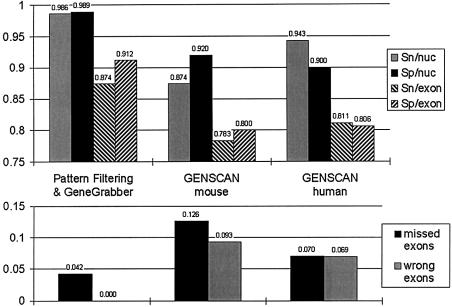
The accurate prediction of higher eukaryotic gene structures and regulatory elements directly from genomic sequences is an important early step in the understanding of newly assembled contigs and finished genomes. As more new genomes are sequenced, comparative approaches are becoming increasingly practical and valuable for predicting genes and regulatory elements. We demonstrate the effectiveness of a comparative method called pattern filtering; it utilizes synteny between two or more genomic segments for the annotation of genomic sequences. Pattern filtering optimally detects the signatures of conserved functional elements despite the stochastic noise inherent in evolutionary processes, allowing more accurate annotation of gene models. We anticipate that pattern filtering will facilitate sequence annotation and the discovery of new functional elements by the genetics and genomics communities.
Figures
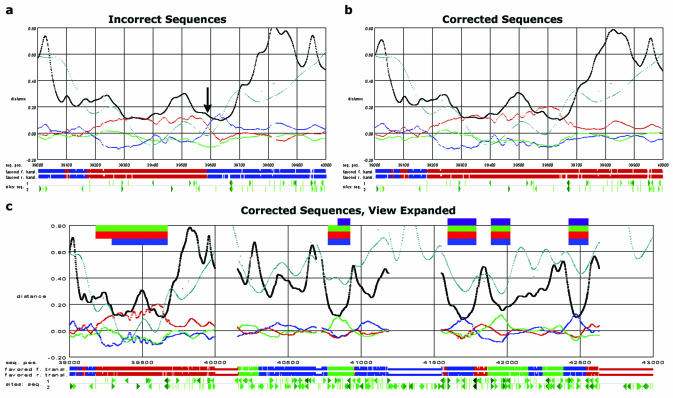
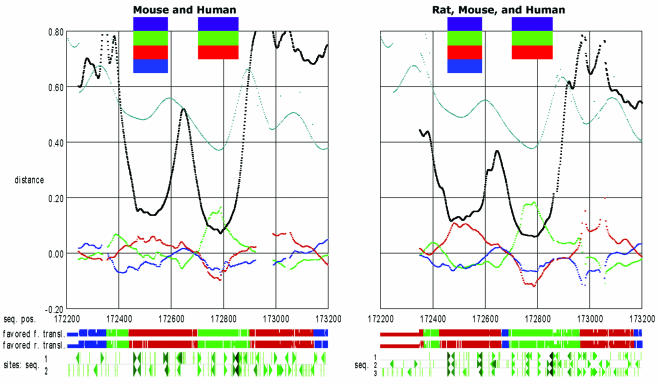
Similar articles
-
DRIMM-Synteny: decomposing genomes into evolutionary conserved segments.Bioinformatics. 2010 Oct 15;26(20):2509-16. doi: 10.1093/bioinformatics/btq465. Epub 2010 Aug 24. Bioinformatics. 2010. PMID: 20736338
-
Rapid detection and curation of conserved DNA via enhanced-BLAT and EvoPrinterHD analysis.BMC Genomics. 2008 Feb 28;9:106. doi: 10.1186/1471-2164-9-106. BMC Genomics. 2008. PMID: 18307801 Free PMC article.
-
How to usefully compare homologous plant genes and chromosomes as DNA sequences.Plant J. 2008 Feb;53(4):661-73. doi: 10.1111/j.1365-313X.2007.03326.x. Plant J. 2008. PMID: 18269575 Review.
-
Numerous potentially functional but non-genic conserved sequences on human chromosome 21.Nature. 2002 Dec 5;420(6915):578-82. doi: 10.1038/nature01251. Nature. 2002. PMID: 12466853
-
Computational identification of transcriptional regulatory elements in DNA sequence.Nucleic Acids Res. 2006 Jul 19;34(12):3585-98. doi: 10.1093/nar/gkl372. Print 2006. Nucleic Acids Res. 2006. PMID: 16855295 Free PMC article. Review.
References
-
- Stormo G. (2000) Gene-finding approaches for eukayotes. Genome Res., 10, 394–397. - PubMed
-
- Claverie J.-M. (1997) Computational methods for the identification of genes in vertebrate genomic sequences. Hum. Mol. Genet., 6, 1735–1744. - PubMed
-
- Fickett J.W. (1996) Finding genes by computer: the state of the art. Trends Genet., 12, 316–320. - PubMed
-
- Haussler D. (1998) Computational genefinding. Trends Guide Bioinformatics (Suppl.), 12–15.
-
- Burge C.B. and Karlin,S. (1998) Finding the genes in genomic DNA. Curr. Opin. Struct. Biol., 8, 346–354. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
