

Identification and characterization of multi-species conserved sequences
- PMID: 14656959
- PMCID: PMC403793
- DOI: 10.1101/gr.1602203
Identification and characterization of multi-species conserved sequences
Abstract
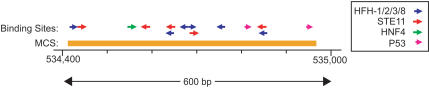
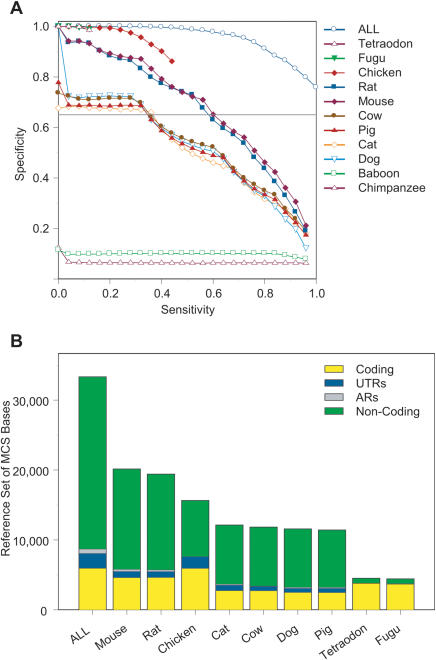
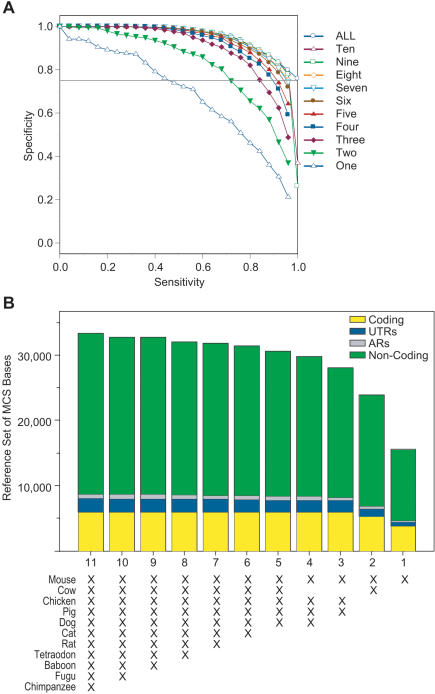
Comparative sequence analysis has become an essential component of studies aiming to elucidate genome function. The increasing availability of genomic sequences from multiple vertebrates is creating the need for computational methods that can detect highly conserved regions in a robust fashion. Towards that end, we are developing approaches for identifying sequences that are conserved across multiple species; we call these "Multi-species Conserved Sequences" (or MCSs). Here we report two strategies for MCS identification, demonstrating their ability to detect virtually all known actively conserved sequences (specifically, coding sequences) but very little neutrally evolving sequence (specifically, ancestral repeats). Importantly, we find that a substantial fraction of the bases within MCSs (approximately 70%) resides within non-coding regions; thus, the majority of sequences conserved across multiple vertebrate species has no known function. Initial characterization of these MCSs has revealed sequences that correspond to clusters of transcription factor-binding sites, non-coding RNA transcripts, and other candidate functional elements. Finally, the ability to detect MCSs represents a valuable metric for assessing the relative contribution of a species' sequence to identifying genomic regions of interest, and our results indicate that the currently available genome sequences are insufficient for the comprehensive identification of MCSs in the human genome.
Figures
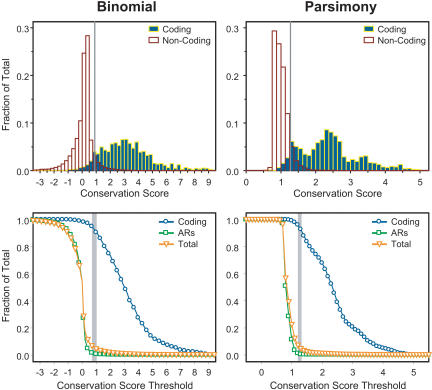
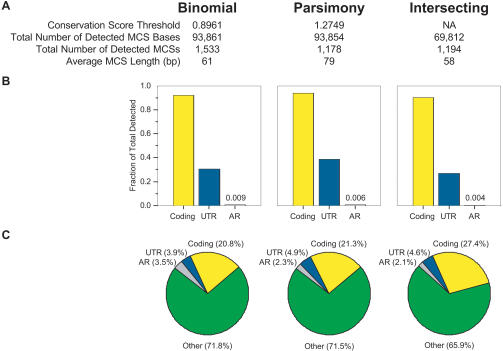
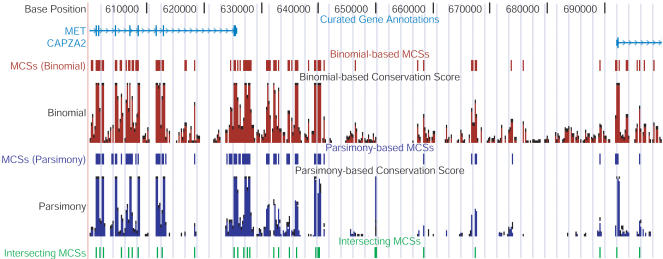
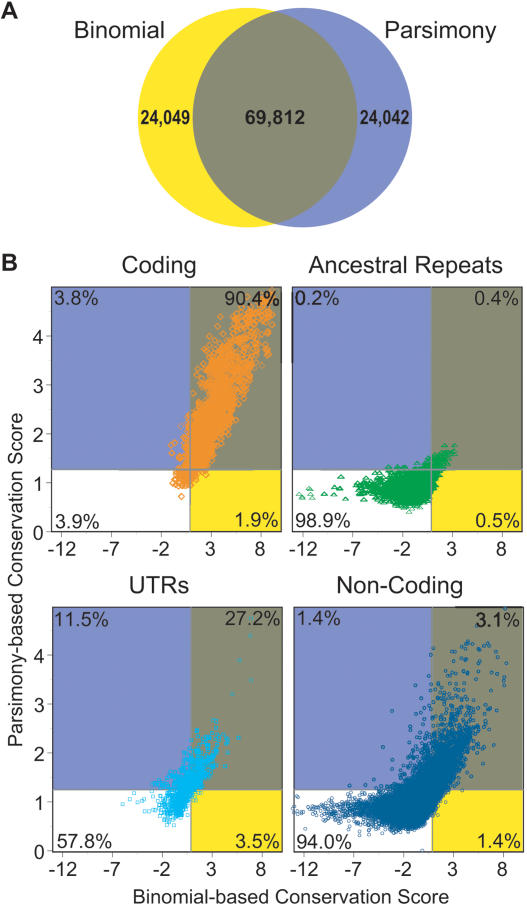
References
-
- Akker, S.A., Smith, P.J., and Chew, S.L. 2001. Nuclear post-transcriptional control of gene expression. J. Mol. Endocrinol. 27: 123-131. - PubMed
-
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. - PubMed
-
- Aparicio, S., Chapman, J., Stupka, E., Putnam, N., Chia, J.M., Dehal, P., Christoffels, A., Rash, S., Hoon, S., Smit, A., et al. 2002. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297: 1301-1310. - PubMed
-
- Bailey, L. and Elkan, C. 1995. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Machine Learning 21: 51-80.
WEB SITE REFERENCES
-
- http://www.nisc.nih.gov; NIH Intramural Sequencing Center (NISC) home page.
-
- http://www.nisc.nih.gov/data; Supplementary data, including annotated sequence for the studies reported here and supplemental tables.
-
- http://genome.ucsc.edu; UC Santa Cruz Genome Browser home page, including the multi-species “zoo browser.”
-
- http://bio.cs.washington.edu; Computational Molecular Biology Group (University of Washington, Computer Science & Engineering) home page.
-
- http://genome.gov/ENCODE; ENCODE project home page.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous