

A genome-wide survey of human pseudogenes
- PMID: 14656963
- PMCID: PMC403797
- DOI: 10.1101/gr.1455503
A genome-wide survey of human pseudogenes
Abstract
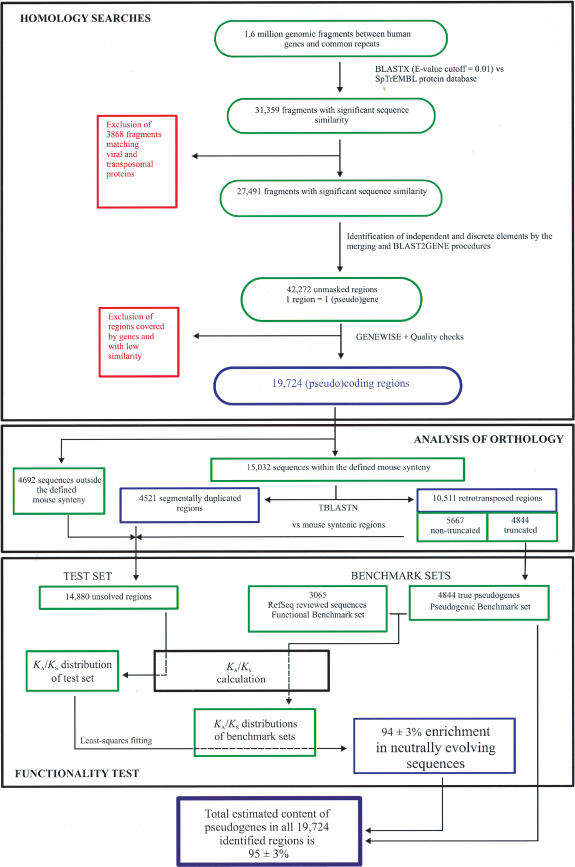
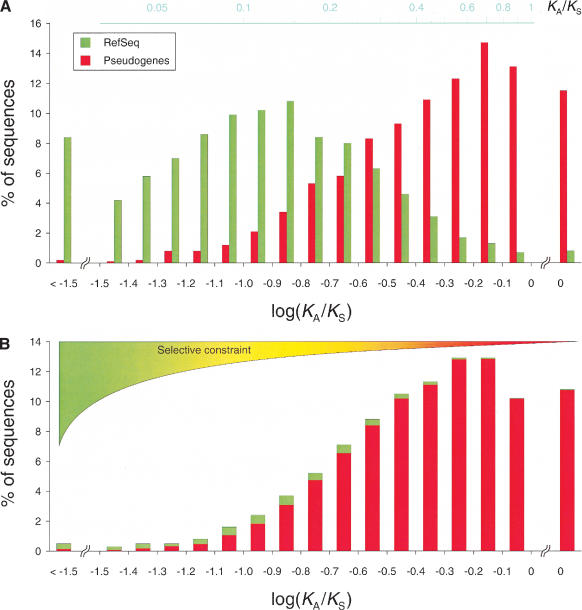
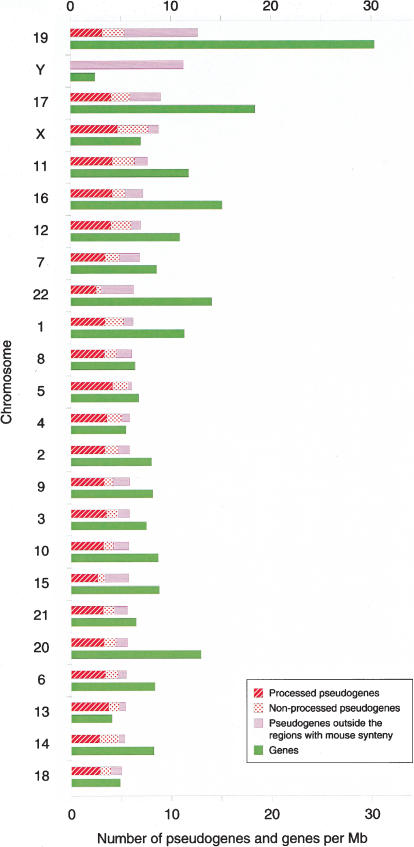
We screened all intergenic regions in the human genome to identify pseudogenes with a combination of homology searches and a functionality test using the ratio of silent to replacement nucleotide substitutions (KA/KS). We identified 19,724 regions of which 95% +/- 3% are estimated to evolve neutrally and thus are likely to encode pseudogenes. Half of these have no detectable truncation in their pseudocoding regions and therefore are not identifiable by methods that require the presence of truncations to prove nonfunctionality. A comparative analysis with the mouse genome showed that 70% of these pseudogenes have a retrotranspositional origin (processed), and the rest arose by segmental duplication (nonprocessed). Although the spread of both types of pseudogenes correlates with chromosome size, nonprocessed pseudogenes appear to be enriched in regions with high gene density. It is likely that the human pseudogenes identified here represent only a small fraction of the total, which probably exceeds the number of genes.
Figures
References
-
- Birney, E. and Durbin, R. 1997. Dynamite: A flexible code generating language for dynamic programming methods used in sequence comparison. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5: 56-64. - PubMed
-
- Brosius, J. 1999. RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene 238: 115-134. - PubMed
-
- Bustamante, C.D., Nielsen, R., and Hartl, D.L. 2002. A maximum likelihood method for analyzing pseudogene evolution: Implications for silent site evolution in humans and rodents. Mol. Biol. Evol. 19: 110-117. - PubMed
WEB SITE REFERENCES
-
- http://www.bork.embl-heidelberg.de/Docu/Human_Pseudogenes/; authors' Web site.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources