

Carbon monoxide protects against liver failure through nitric oxide-induced heme oxygenase 1
- PMID: 14657222
- PMCID: PMC2194127
- DOI: 10.1084/jem.20031003
Carbon monoxide protects against liver failure through nitric oxide-induced heme oxygenase 1
Abstract
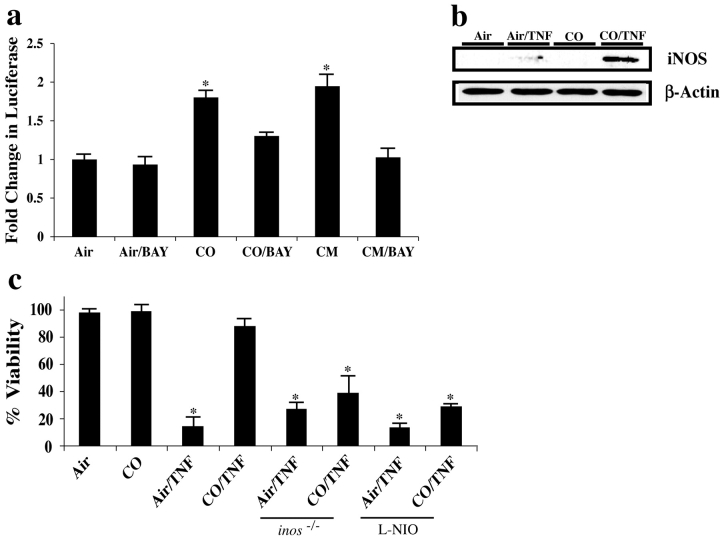
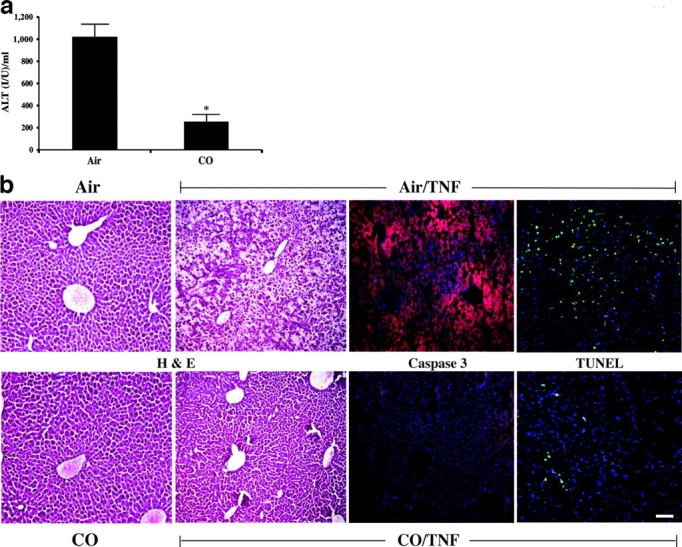
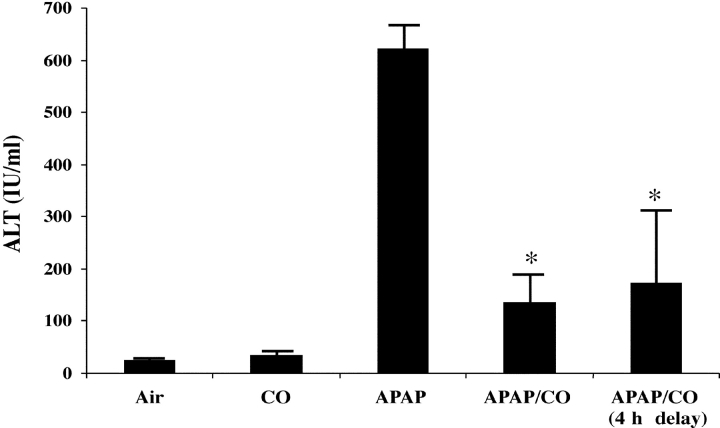
Carbon monoxide (CO) and nitric oxide (NO) each have mechanistically unique roles in various inflammatory disorders. Although it is known that CO can induce production of NO and that NO can induce expression of the cytoprotective enzyme heme oxygenase 1 (HO-1), there is no information whether the protective effect of CO ever requires NO production or whether either gas must induce expression of HO-1 to exert its functional effects. Using in vitro and in vivo models of tumor necrosis factor alpha-induced hepatocyte cell death in mice, we find that activation of nuclear factor kappaB and increased expression of inducible NO are required for the protective effects of CO, whereas the protective effects of NO require up-regulation of HO-1 expression. When protection from cell death is initiated by CO, NO production and HO-1 activity are each required for the protective effect showing for the first time an essential synergy between these two molecules in tandem providing potent cytoprotection.
Figures







Similar articles
-
Galantamine and carbon monoxide protect brain microvascular endothelial cells by heme oxygenase-1 induction.Biochem Biophys Res Commun. 2008 Mar 14;367(3):674-9. doi: 10.1016/j.bbrc.2007.12.152. Epub 2008 Jan 2. Biochem Biophys Res Commun. 2008. PMID: 18174021
-
Carbon monoxide and nitric oxide protect against tumor necrosis factor-alpha-induced apoptosis in osteoblasts: HO-1 is necessary to mediate the protection.Clin Chim Acta. 2006 Mar;365(1-2):270-8. doi: 10.1016/j.cca.2005.09.011. Epub 2005 Oct 19. Clin Chim Acta. 2006. PMID: 16242122
-
Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver.FASEB J. 2004 May;18(7):854-6. doi: 10.1096/fj.03-0643fje. Epub 2004 Mar 4. FASEB J. 2004. PMID: 15001560
-
[Carbon monoxide--a toxic gas and ...a signal molecule with therapeutic potential].Lakartidningen. 2005 Feb 28-Mar 6;102(9):642, 645-7. Lakartidningen. 2005. PMID: 15804036 Review. Swedish.
-
Modulation of endothelial cell apoptosis by heme oxygenase-1-derived carbon monoxide.Antioxid Redox Signal. 2002 Apr;4(2):321-9. doi: 10.1089/152308602753666370. Antioxid Redox Signal. 2002. PMID: 12006183 Review.
Cited by
-
Regulation of Endothelial and Vascular Functions by Carbon Monoxide via Crosstalk With Nitric Oxide.Front Cardiovasc Med. 2021 Apr 12;8:649630. doi: 10.3389/fcvm.2021.649630. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 33912601 Free PMC article. Review.
-
Heme oxygenase-1 deficiency promotes the development of necrotizing enterocolitis-like intestinal injury in a newborn mouse model.Am J Physiol Gastrointest Liver Physiol. 2013 Jun 1;304(11):G991-G1001. doi: 10.1152/ajpgi.00363.2012. Epub 2013 Apr 11. Am J Physiol Gastrointest Liver Physiol. 2013. PMID: 23578787 Free PMC article.
-
Administration of a CO-releasing molecule induces late preconditioning against myocardial infarction.J Mol Cell Cardiol. 2005 Jan;38(1):127-34. doi: 10.1016/j.yjmcc.2004.10.006. Epub 2004 Dec 8. J Mol Cell Cardiol. 2005. PMID: 15623429 Free PMC article.
-
The Role of Gasotransmitter-Dependent Signaling Mechanisms in Apoptotic Cell Death in Cardiovascular, Rheumatic, Kidney, and Neurodegenerative Diseases and Mental Disorders.Int J Mol Sci. 2023 Mar 23;24(7):6014. doi: 10.3390/ijms24076014. Int J Mol Sci. 2023. PMID: 37046987 Free PMC article. Review.
-
Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1.Am J Respir Crit Care Med. 2008 Jun 1;177(11):1223-32. doi: 10.1164/rccm.200708-1265OC. Epub 2008 Mar 20. Am J Respir Crit Care Med. 2008. PMID: 18356564 Free PMC article.
References
-
- Saavedra, J.E., T.R. Billiar, D.L. Williams, Y.M. Kim, S.C. Watkins, and L.K. Keefer. 1997. Targeting nitric oxide (NO) delivery in vivo. Design of a liver-selective NO donor prodrug that blocks tumor necrosis factor-alpha-induced apoptosis and toxicity in the liver. J. Med. Chem. 40:1947–1954. - PubMed
-
- Mojena, M., S. Hortelano, A. Castrillo, M.J. Diaz-Guerra, M.J. Garcia-Barchino, G.T. Saez, and L. Bosca. 2001. Protection by nitric oxide against liver inflammatory injury in animals carrying a nitric oxide synthase-2 transgene. FASEB J. 15:583–585. - PubMed
-
- Otterbein, L., S.L. Sylvester, and A.M. Choi. 1995. Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am. J. Respir. Cell Mol. Biol. 13:595–601. - PubMed
-
- Tamion, F., V. Richard, Y. Lacoume, and C. Thuillez. 2002. Intestinal preconditioning prevents systemic inflammatory response in hemorrhagic shock. Role of HO-1. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G408–G414. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
