

Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death
- PMID: 14663141
- PMCID: PMC307684
- DOI: 10.1073/pnas.2534856100
Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death
Erratum in
- Proc Natl Acad Sci U S A. 2004 Feb 3;101(5):1426
Abstract
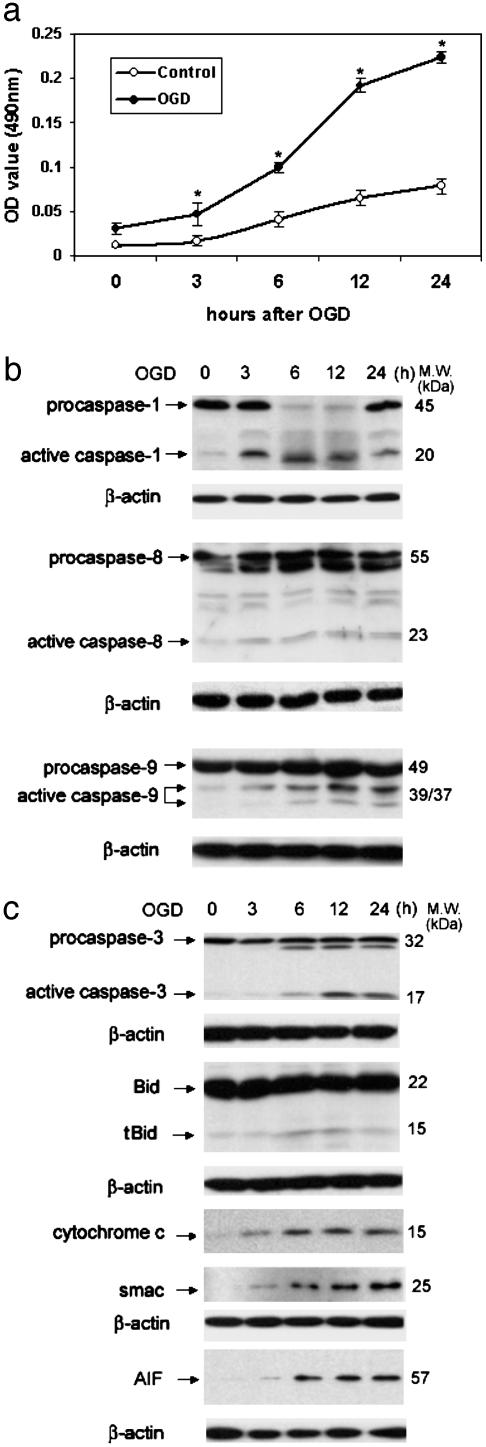
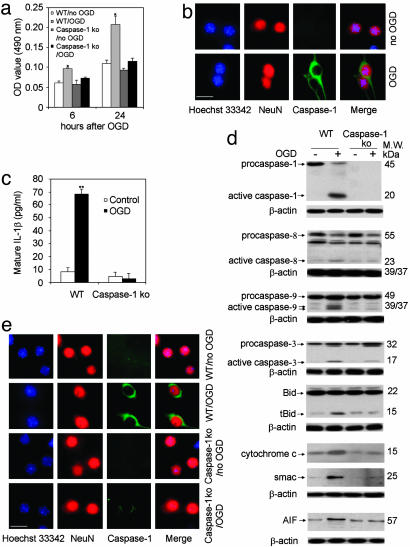
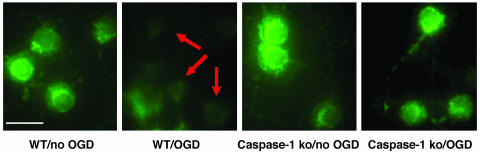
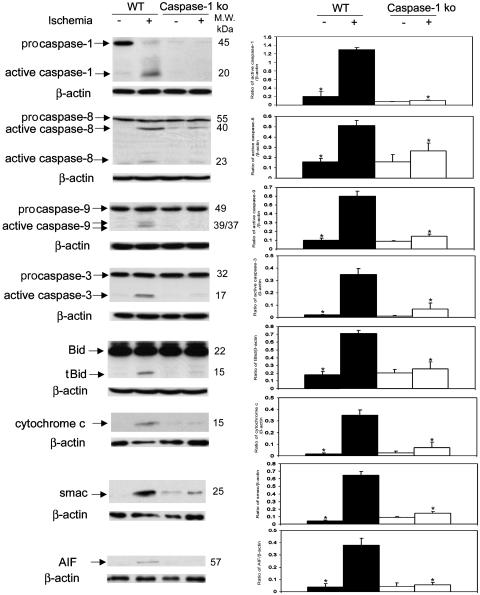
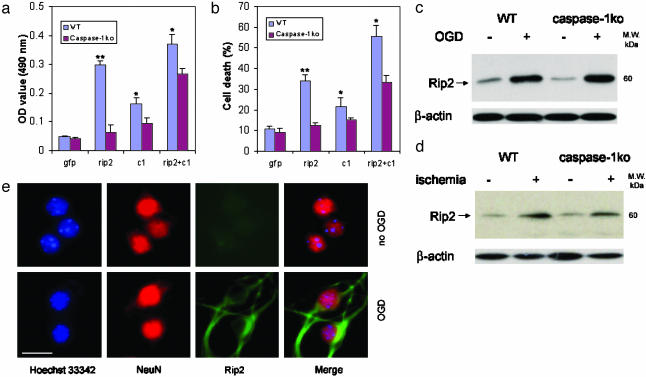
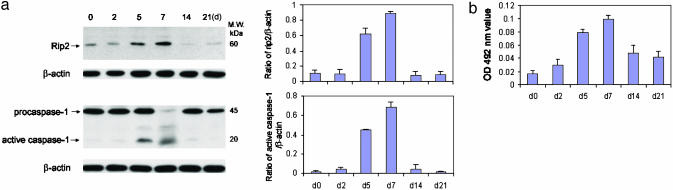
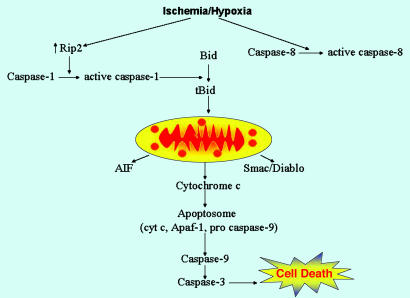
Caspase-1 plays a key role in inflammatory pathways by processing pro-IL-1beta into the active cytokine mature IL-1beta. Given its sequence similarity with the Caenorhabditis elegans cell death gene ced-3,it has long been speculated that caspase-1 may also play a role in cell death. However, an unequivocal role for caspase-1 in cell death has been questioned, and not definitively demonstrated. Furthermore, if caspase-1 does play a role in cell death, its position in the apoptotic hierarchy has not been clearly defined. Previous studies have shown that caspase-1 knockout (KO) mice and transgenic mice expressing a dominant-negative caspase-1 construct are resistant to ischemic brain injury. We provide direct evidence that caspase-1 plays a key role in neuronal cell death and that caspase-1 is an apical activator of the cell death pathway in the premitochondrial collapse stage. Furthermore, we demonstrate that Rip2/Cardiak/Rick is a stress-inducible upstream modulator of pro-caspase-1 apoptotic activation. We provide evidence that Bid cleavage appears to be an important downstream effector of caspase-1-mediated cell death. Our data demonstrate that caspase-1 is an apical mediator of neuronal cell death during in vitro hypoxia, and confirmed in vivo in ischemia, and provide insights into the sequence of events involved in this pathological cell death process.
Figures
References
-
- Thornberry, N. A., Rano, T. A., Peterson, E. P., Rasper, D. M., Timkey, T., Garcia-Calvo, M., Houtzager, V. M., Nordstrom, P. A., Roy, S., Vaillancourt, J. P., et al. (1997) J. Biol. Chem. 272, 17907-17911. - PubMed
-
- Shi, Y. (2002) Mol. Cell 9, 459-470. - PubMed
-
- Wang, S., Miura, M., Jung, Y. K., Zhu, H., Li, E. & Yuan, J. (1998) Cell 92, 501-509. - PubMed
-
- Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A. & Yuan, J. (2000) Nature 403, 98-103. - PubMed
-
- Li, M., Ona, V. O., Guegan, C., Chen, M., Jackson-Lewis, V., Andrews, L. J., Olszewski, A. J., Stieg, P. E., Lee, J. P., Przedborski, S. & Friedlander, R. M. (2000) Science 288, 335-339. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
