

Molecular basis of bacterial outer membrane permeability revisited
- PMID: 14665678
- PMCID: PMC309051
- DOI: 10.1128/MMBR.67.4.593-656.2003
Molecular basis of bacterial outer membrane permeability revisited
Abstract
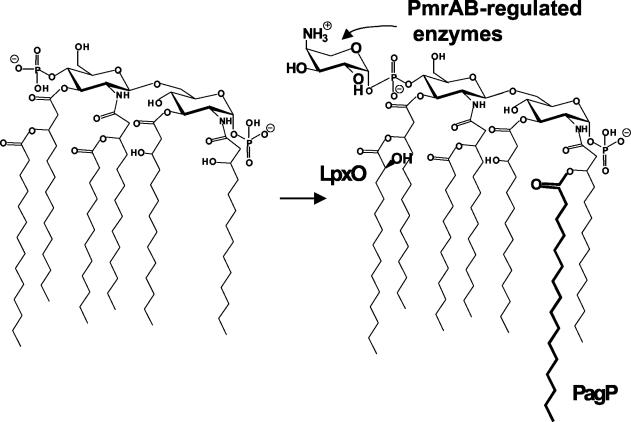
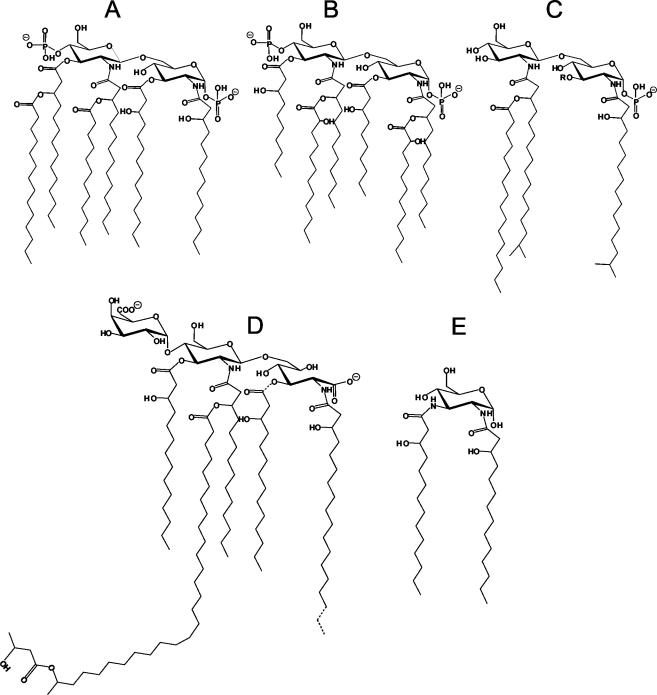
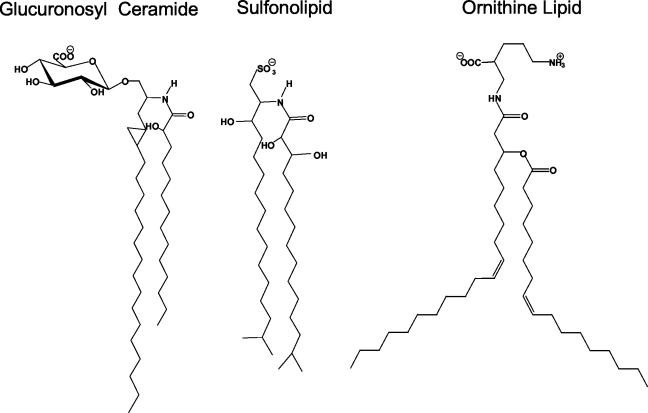
Gram-negative bacteria characteristically are surrounded by an additional membrane layer, the outer membrane. Although outer membrane components often play important roles in the interaction of symbiotic or pathogenic bacteria with their host organisms, the major role of this membrane must usually be to serve as a permeability barrier to prevent the entry of noxious compounds and at the same time to allow the influx of nutrient molecules. This review summarizes the development in the field since our previous review (H. Nikaido and M. Vaara, Microbiol. Rev. 49:1-32, 1985) was published. With the discovery of protein channels, structural knowledge enables us to understand in molecular detail how porins, specific channels, TonB-linked receptors, and other proteins function. We are now beginning to see how the export of large proteins occurs across the outer membrane. With our knowledge of the lipopolysaccharide-phospholipid asymmetric bilayer of the outer membrane, we are finally beginning to understand how this bilayer can retard the entry of lipophilic compounds, owing to our increasing knowledge about the chemistry of lipopolysaccharide from diverse organisms and the way in which lipopolysaccharide structure is modified by environmental conditions.
Figures

References
-
- Abbanat, D. R., W. Godchaux III, and E. R. Leadbetter. 1988. Surface-induced synthesis of new sulfonolipids in the gliding bacterium Cytophaga johnsonae. Arch. Microbiol. 149:358-364.
-
- Abbanat, D. R., E. R. Leadbetter, W. Godchaux III, and A. Escher. 1986. Sulfonolipids are molecular determinants of gliding motility. Nature 324:367-369.
-
- Abergel, C., E. Bouveret, J. M. Claverie, K. Brown, A. Rigal, C. Lazdunski, and H. Benedetti. 1999. Structure of the Escherichia coli TolB protein determined by MAD methods at 1.95 Å resolution. Struct. Fold Des. 7:1291-1300. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
