

Mosaic evolution of the severe acute respiratory syndrome coronavirus
- PMID: 14671089
- PMCID: PMC303383
- DOI: 10.1128/jvi.78.1.76-82.2004
Mosaic evolution of the severe acute respiratory syndrome coronavirus
Abstract
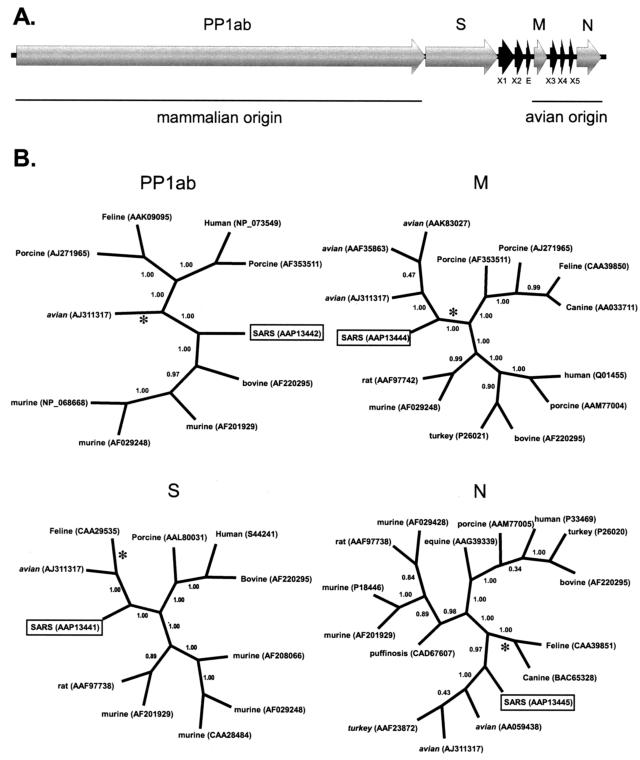
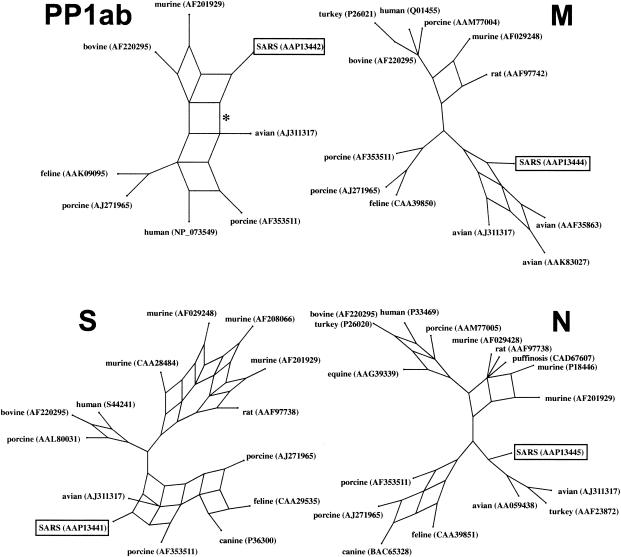
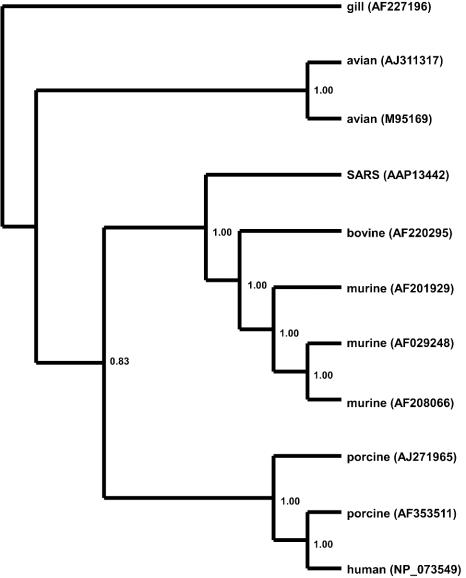
Severe acute respiratory syndrome (SARS) is a deadly form of pneumonia caused by a novel coronavirus, a viral family responsible for mild respiratory tract infections in a wide variety of animals including humans, pigs, cows, mice, cats, and birds. Analyses to date have been unable to identify the precise origin of the SARS coronavirus. We used Bayesian, neighbor-joining, and split decomposition phylogenetic techniques on the SARS virus replicase, surface spike, matrix, and nucleocapsid proteins to reveal the evolutionary origin of this recently emerging infectious agent. The analyses support a mammalian-like origin for the replicase protein, an avian-like origin for the matrix and nucleocapsid proteins, and a mammalian-avian mosaic origin for the host-determining spike protein. A bootscan recombination analysis of the spike gene revealed high nucleotide identity between the SARS virus and a feline infectious peritonitis virus throughout the gene, except for a 200- base-pair region of high identity to an avian sequence. These data support the phylogenetic analyses and suggest a possible past recombination event between mammalian-like and avian-like parent viruses. This event occurred near a region that has been implicated to be the human receptor binding site and may have been directly responsible for the switch of host of the SARS coronavirus from animals to humans.
Figures
References
-
- Alejska, M., A. Kurzynska-Kokorniak, M. Broda, R. Kierzek, and M. Figlerowicz. 2001. How RNA viruses exchange their genetic material. Acta Biochim. Pol. 48:391-407. - PubMed
-
- Anand, K., J. Ziebuhr, P. Wadhwani, J. R. Mesters, and R. Hilgenfeld. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763-1767. - PubMed
-
- Chang, S. H., J. L. Bae, T. J. Kang, J. Kim, G. H. Chung, C. W. Lim, H. Laude, M. S. Yang, and Y. S. Jang. 2002. Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol. Cell 14:295-299. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
