

Whole-genome discovery of transcription factor binding sites by network-level conservation
- PMID: 14672978
- PMCID: PMC314286
- DOI: 10.1101/gr.1739204
Whole-genome discovery of transcription factor binding sites by network-level conservation
Abstract
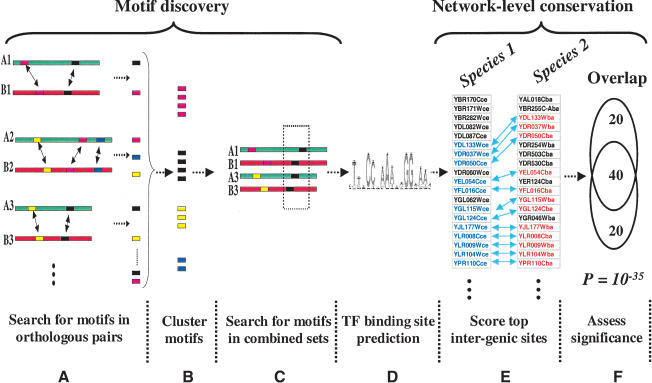
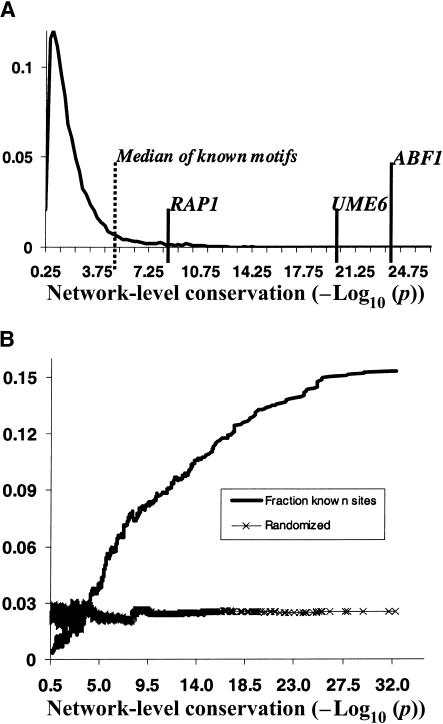
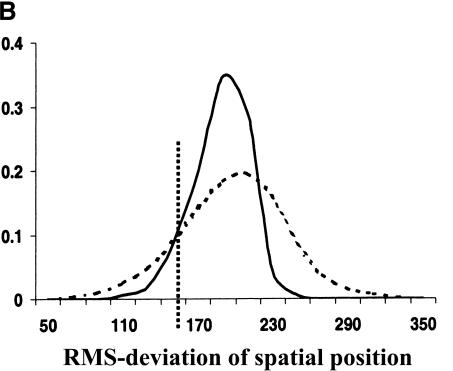
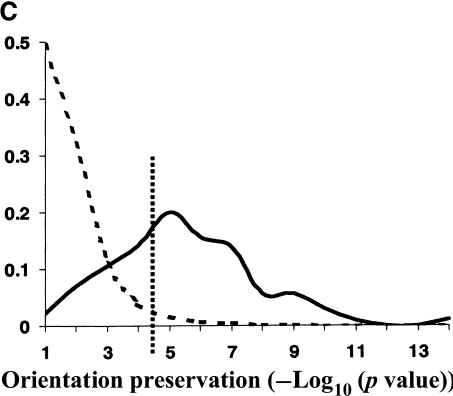
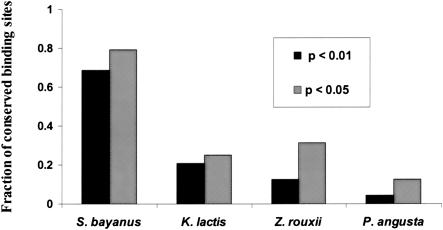
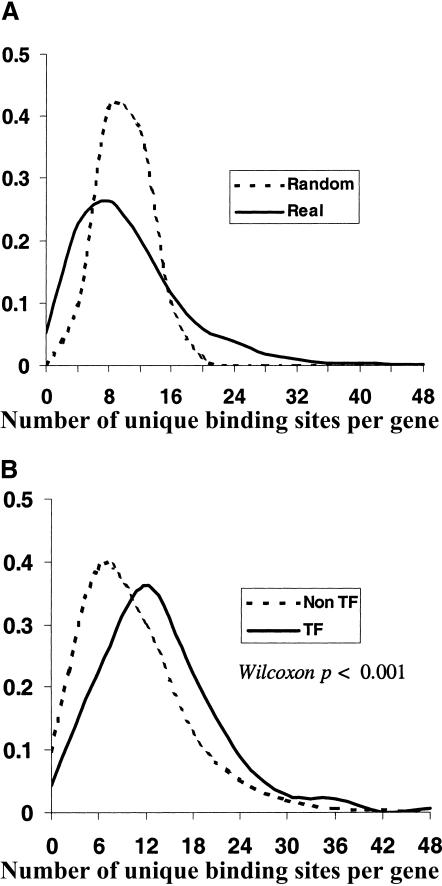
Comprehensive identification of DNA cis-regulatory elements is crucial for a predictive understanding of transcriptional network dynamics. Strong evidence suggests that these DNA sequence motifs are highly conserved between related species, reflecting strong selection on the network of regulatory interactions that underlie common cellular behavior. Here, we exploit a systems-level aspect of this conservation-the network-level topology of these interactions-to map transcription factor (TF) binding sites on a genomic scale. Using network-level conservation as a constraint, our algorithm finds 71% of known TF binding sites in the yeast Saccharomyces cerevisiae, using only 12% of the sequence of a phylogenetic neighbor. Most of the novel predicted motifs show strong features of known TF binding sites, such as functional category and/or expression profile coherence of their corresponding genes. Network-level conservation should provide a powerful constraint for the systematic mapping of TF binding sites in the larger genomes of higher eukaryotes.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous