

Diversity of regulatory CD4+T cells controlling distinct organ-specific autoimmune diseases
- PMID: 14673094
- PMCID: PMC307649
- DOI: 10.1073/pnas.2636971100
Diversity of regulatory CD4+T cells controlling distinct organ-specific autoimmune diseases
Erratum in
- Proc Natl Acad Sci U S A. 2004 Mar 23;101(12):4331
Abstract
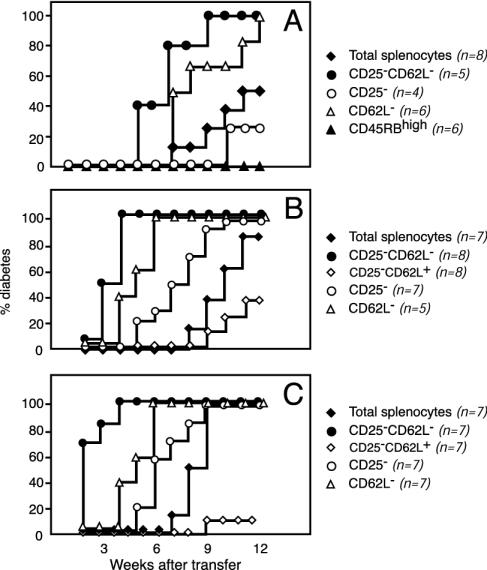
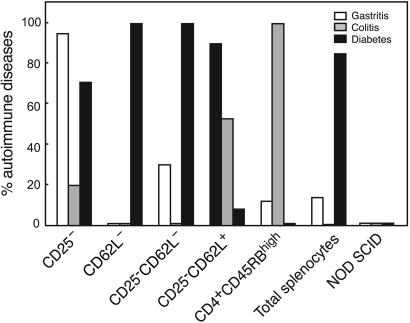
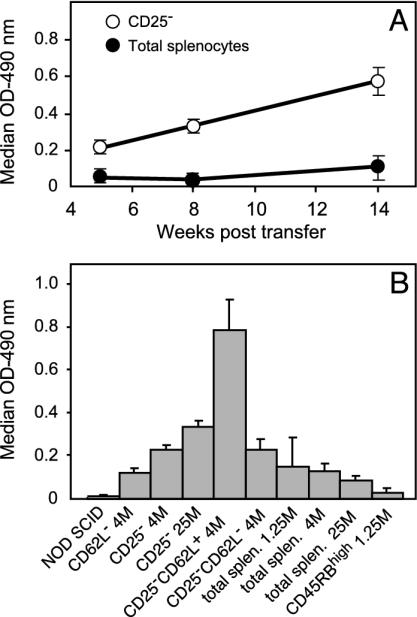
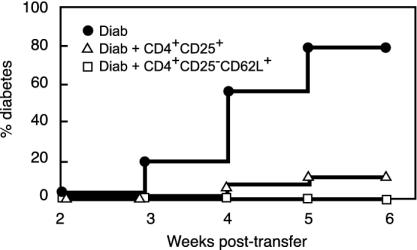
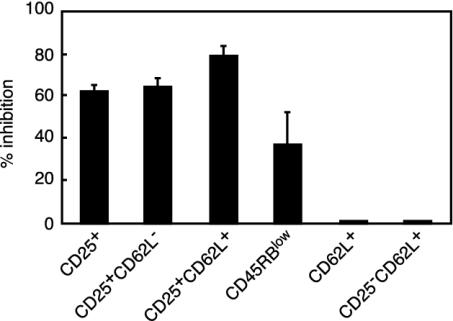
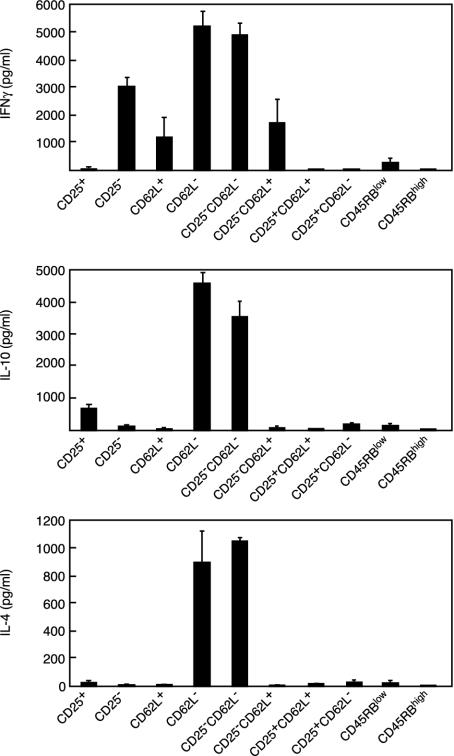
Depletion of selected regulatory CD4+ T cell subsets induces the spontaneous onset of various immune or autoimmune disorders. It is not clear, however, whether a given subset, notably CD4+CD25+ regulatory T cells, protects from a wide spectrum of immune disorders, or whether specialized subsets of regulatory T cells control each given disease or group of diseases. We report here, using diabetes prone nonobese diabetic (NOD) mice, that depending on the regulatory T cells that are depleted, i.e., CD25+, CD62L+, or CD45RB(low), distinct immune diseases appear after transfer into NOD severe combined immunodeficiency (SCID) recipients. Thus, reconstitution of NOD SCID mice with CD25- T cells induces major gastritis and late-onset diabetes, but no or mild colitis. Reconstitution with CD62L- T cells induces fulminant diabetes with no colitis or gastritis. Reconstitution with CD45RB(high) T cells induces major colitis with wasting disease and no or very moderate gastritis and diabetes. Major differences among the three regulatory T cell subsets are also seen in vitro. The bulk of suppressor cells inhibiting the proliferation of CD4+CD25- T cells in coculture is concentrated within the CD25+ but not the CD62L+ or CD45RB(low) T cell subsets. Similarly, cytokine production patterns are significantly different for each regulatory T cell subset. Collectively, these data point to the diversity and organ selectivity of regulatory T cells controlling distinct autoimmune diseases whatever the underlying mechanisms.
Figures
References
-
- Shevach, E. M. (2002) Nat. Rev. Immunol. 2, 389-400. - PubMed
-
- Bach, J. F. (2003) Nat. Rev. Immunol. 3, 189-198. - PubMed
-
- Powrie, F., Leach, M. W., Mauze, S., Caddle, L. B. & Coffman, R. L. (1993) Int. Immunol. 5, 1461-1471. - PubMed
-
- Herbelin, A., Gombert, J. M., Lepault, F., Bach, J. F. & Chatenoud, L. (1998) J. Immunol. 161, 2620-2628. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
