

Network component analysis: reconstruction of regulatory signals in biological systems
- PMID: 14673099
- PMCID: PMC307600
- DOI: 10.1073/pnas.2136632100
Network component analysis: reconstruction of regulatory signals in biological systems
Abstract
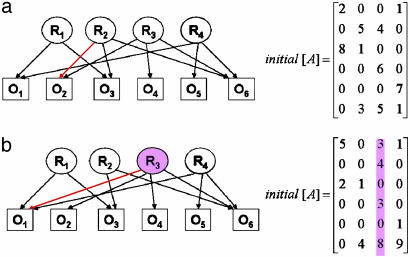
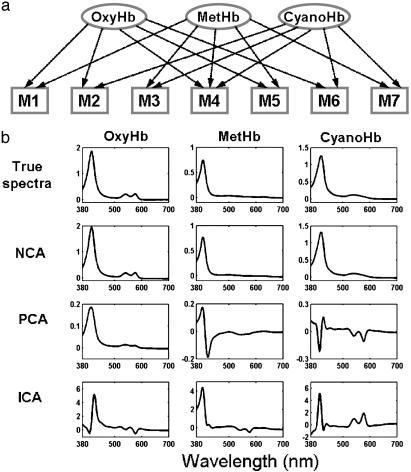
High-dimensional data sets generated by high-throughput technologies, such as DNA microarray, are often the outputs of complex networked systems driven by hidden regulatory signals. Traditional statistical methods for computing low-dimensional or hidden representations of these data sets, such as principal component analysis and independent component analysis, ignore the underlying network structures and provide decompositions based purely on a priori statistical constraints on the computed component signals. The resulting decomposition thus provides a phenomenological model for the observed data and does not necessarily contain physically or biologically meaningful signals. Here, we develop a method, called network component analysis, for uncovering hidden regulatory signals from outputs of networked systems, when only a partial knowledge of the underlying network topology is available. The a priori network structure information is first tested for compliance with a set of identifiability criteria. For networks that satisfy the criteria, the signals from the regulatory nodes and their strengths of influence on each output node can be faithfully reconstructed. This method is first validated experimentally by using the absorbance spectra of a network of various hemoglobin species. The method is then applied to microarray data generated from yeast Saccharamyces cerevisiae and the activities of various transcription factors during cell cycle are reconstructed by using recently discovered connectivity information for the underlying transcriptional regulatory networks.
Figures


References
-
- Schena, M., Shalon, D., Davis, R. W. & Brown, P. O. (1995) Science 270, 467–470. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
