

Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction
- PMID: 14679178
- PMCID: PMC297000
- DOI: 10.1172/JCI19774
Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction
Abstract
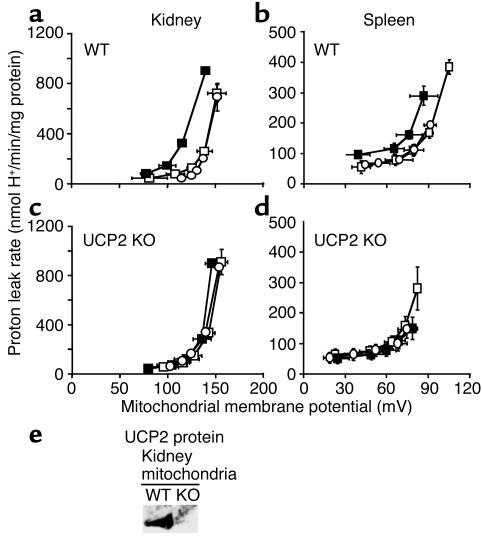
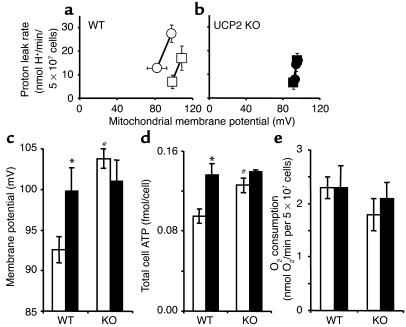
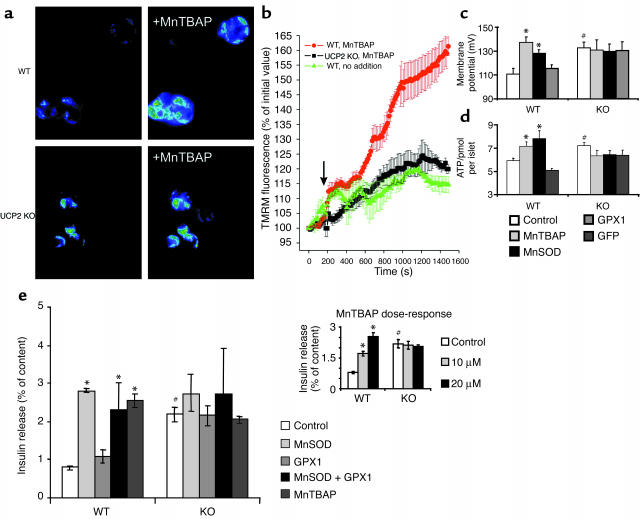
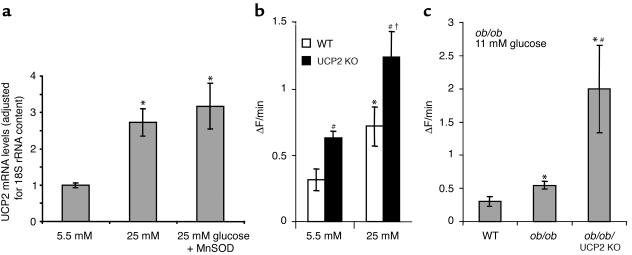
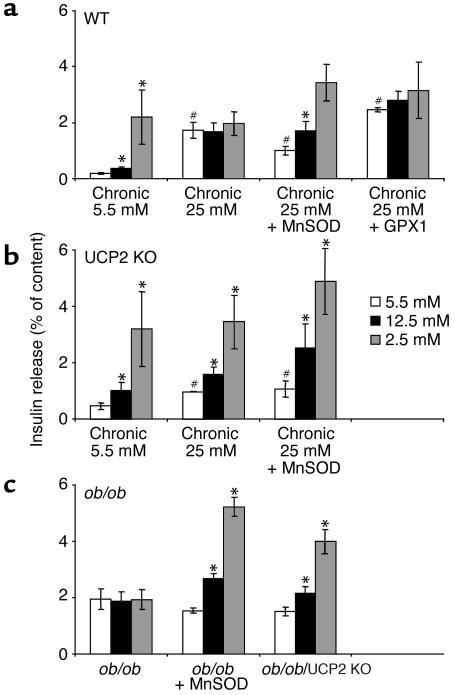
Failure to secrete adequate amounts of insulin in response to increasing concentrations of glucose is an important feature of type 2 diabetes. The mechanism for loss of glucose responsiveness is unknown. Uncoupling protein 2 (UCP2), by virtue of its mitochondrial proton leak activity and consequent negative effect on ATP production, impairs glucose-stimulated insulin secretion. Of interest, it has recently been shown that superoxide, when added to isolated mitochondria, activates UCP2-mediated proton leak. Since obesity and chronic hyperglycemia increase mitochondrial superoxide production, as well as UCP2 expression in pancreatic beta cells, a superoxide-UCP2 pathway could contribute importantly to obesity- and hyperglycemia-induced beta cell dysfunction. This study demonstrates that endogenously produced mitochondrial superoxide activates UCP2-mediated proton leak, thus lowering ATP levels and impairing glucose-stimulated insulin secretion. Furthermore, hyperglycemia- and obesity-induced loss of glucose responsiveness is prevented by reduction of mitochondrial superoxide production or gene knockout of UCP2. Importantly, reduction of superoxide has no beneficial effect in the absence of UCP2, and superoxide levels are increased further in the absence of UCP2, demonstrating that the adverse effects of superoxide on beta cell glucose sensing are caused by activation of UCP2. Therefore, superoxide-mediated activation of UCP2 could play an important role in the pathogenesis of beta cell dysfunction and type 2 diabetes.
Figures
Comment in
-
A radical explanation for glucose-induced beta cell dysfunction.J Clin Invest. 2003 Dec;112(12):1788-90. doi: 10.1172/JCI20501. J Clin Invest. 2003. PMID: 14679173 Free PMC article.
References
-
- Matthews DR, Clark A. B-cell defects and pancreatic abnormalities in non-insulin-dependent diabetes mellitus. In Textbook of diabetes. JC Pickup and G Williams, editors Blackwell Science Oxford, United Kingdom. 1997;21:21–16.
-
- Poitout V, Robertson RP. Minireview. Secondary beta-cell failure in type 2 diabetes: a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342. - PubMed
-
- Robertson, R.P., et al. 2000. Glucose toxicity of the beta cell: cellular and molecular mechanisms. In Diabetes mellitus. A fundamental and clinical text. D. Le Roith, S. Taylor, and J.M. Olefsky, editors. Lippincott Williams & Wilkins. Philadelphia, Pennsylvania, USA. 125–132.
-
- Jaburek M, et al. Transport function and regulation of mitochondrial uncoupling proteins 2 and 3. J. Biol. Chem. 1999;274:26003–26007. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
