

SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase
- PMID: 14681206
- PMCID: PMC305258
- DOI: 10.1101/gad.1157503
SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase
Abstract
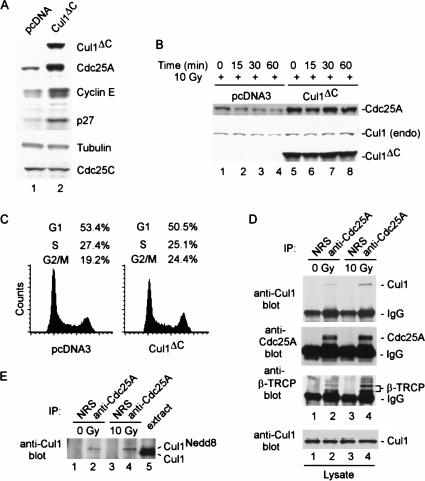
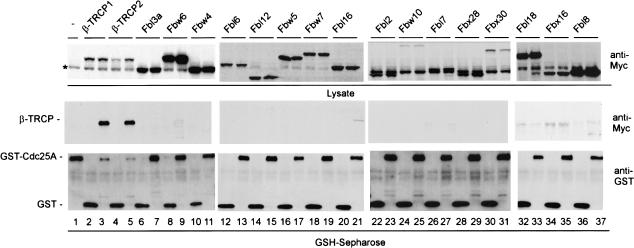
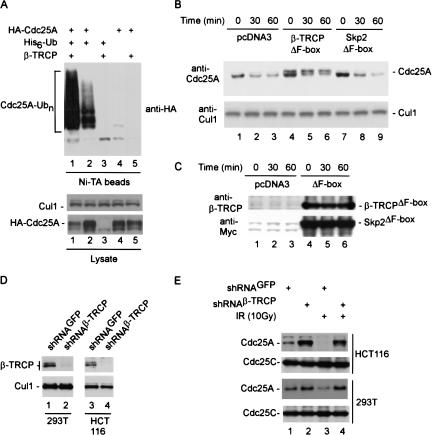
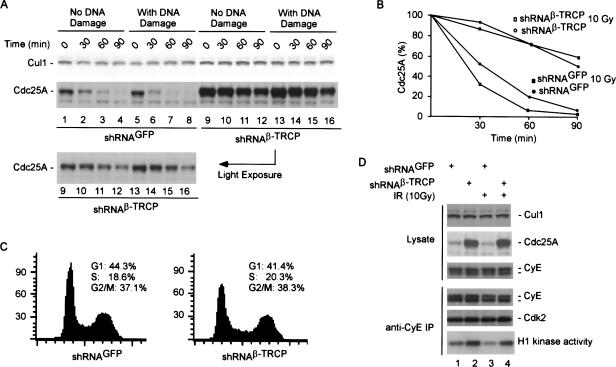
Eukaryotic cells respond to DNA damage and stalled replication forks by activating protein kinase-mediated signaling pathways that promote cell cycle arrest and DNA repair. A central target of the cell cycle arrest program is the Cdc25A protein phosphatase. Cdc25A is required for S-phase entry and dephosphorylates tyrosine-15 phosphorylated Cdk1 (Cdc2) and Cdk2, positive regulators of cell division. Cdc25A is unstable during S-phase and is degraded through the ubiquitin-proteasome pathway, but its turnover is enhanced in response to DNA damage. Although basal and DNA-damage-induced turnover depends on the ATM-Chk2 and ATR-Chk1 pathways, how these kinases engage the ubiquitin ligase machinery is unknown. Here, we demonstrate a requirement for SCFbeta-TRCP in Cdc25A turnover during an unperturbed cell cycle and in response to DNA damage. Depletion of beta-TRCP stabilizes Cdc25A, leading to hyperactive Cdk2 activity. SCFbeta-TRCP promotes Chk1-dependent Cdc25A ubiquitination in vitro, and this involves serine 76, a known Chk1 phosphorylation site. However, recognition of Cdc25A by beta-TRCP occurs via a noncanonical phosphodegron in Cdc25A containing phosphoserine 79 and phosphoserine 82, sites that are not targeted by Chk1. These data indicate that Cdc25A turnover is more complex than previously appreciated and suggest roles for an additional kinase(s) in Chk1-dependent Cdc25A turnover.
Figures
Similar articles
-
Differential roles for checkpoint kinases in DNA damage-dependent degradation of the Cdc25A protein phosphatase.J Biol Chem. 2008 Jul 11;283(28):19322-8. doi: 10.1074/jbc.M802474200. Epub 2008 May 13. J Biol Chem. 2008. PMID: 18480045 Free PMC article.
-
Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage.Nature. 2003 Nov 6;426(6962):87-91. doi: 10.1038/nature02082. Nature. 2003. PMID: 14603323
-
Dual regulation of Cdc25A by Chk1 and p53-ATF3 in DNA replication checkpoint control.J Biol Chem. 2009 Feb 13;284(7):4132-9. doi: 10.1074/jbc.M808118200. Epub 2008 Dec 7. J Biol Chem. 2009. PMID: 19060337
-
Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis.Oncogene. 2004 Mar 15;23(11):2050-6. doi: 10.1038/sj.onc.1207394. Oncogene. 2004. PMID: 15021892 Review.
-
Chk1 versus Cdc25: chking one's levels of cellular proliferation.Cell Cycle. 2004 Nov;3(11):1355-7. doi: 10.4161/cc.3.11.1225. Epub 2004 Nov 10. Cell Cycle. 2004. PMID: 15483403 Review.
Cited by
-
Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway.Mol Cell Biol. 2007 Apr;27(8):3098-108. doi: 10.1128/MCB.02357-06. Epub 2007 Feb 12. Mol Cell Biol. 2007. PMID: 17296736 Free PMC article.
-
Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair.J Nucleic Acids. 2010 Dec 16;2010:592980. doi: 10.4061/2010/592980. J Nucleic Acids. 2010. PMID: 21209706 Free PMC article.
-
SCF(β-TRCP)-mediated degradation of NEDD4 inhibits tumorigenesis through modulating the PTEN/Akt signaling pathway.Oncotarget. 2014 Feb 28;5(4):1026-37. doi: 10.18632/oncotarget.1675. Oncotarget. 2014. PMID: 24657926 Free PMC article.
-
Multisite phosphorylation provides an effective and flexible mechanism for switch-like protein degradation.PLoS One. 2010 Dec 13;5(12):e14029. doi: 10.1371/journal.pone.0014029. PLoS One. 2010. PMID: 21179196 Free PMC article.
-
Targeting the Checkpoint to Kill Cancer Cells.Biomolecules. 2015 Aug 18;5(3):1912-37. doi: 10.3390/biom5031912. Biomolecules. 2015. PMID: 26295265 Free PMC article. Review.
References
-
- Agami R. and Bernards, R. 2000. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 102: 55-66. - PubMed
-
- Bai C., Sen, P., Hofmann, K., Ma, L., Goebl, M., Harper, J.W., and Elledge, S.J. 1996. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86: 263-274. - PubMed
-
- Blasina A., de Weyer, I.V., Laus, M.C., Luyten, W.H., Parker, A.E., and McGowan, C.H. 1999. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr. Biol. 9: 1-10. - PubMed
-
- Deshaies R.J. 1999. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol. 15: 435-467. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous