

Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies
- PMID: 14681476
- PMCID: PMC308741
- DOI: 10.1093/nar/gkh006
Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies
Abstract
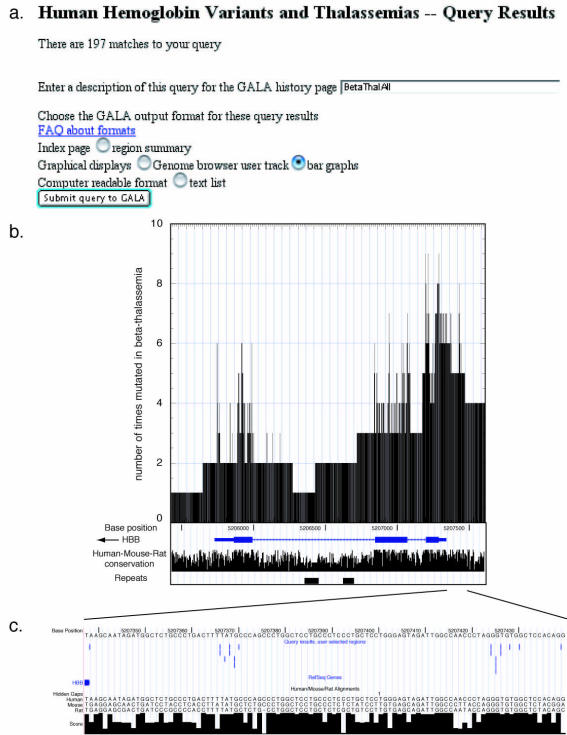
HbVar (http://globin.cse.psu.edu/globin/hbvar/) is a relational database developed by a multi-center academic effort to provide up-to-date and high quality information on the genomic sequence changes leading to hemoglobin variants and all types of thalassemia and hemoglobinopathies. Extensive information is recorded for each variant and mutation, including sequence alterations, biochemical and hematological effects, associated pathology, ethnic occurrence and references. In addition to the regular updates to entries, we report two significant advances: (i) The frequencies for a large number of mutations causing beta-thalassemia in at-risk populations have been extracted from the published literature and made available for the user to query upon. (ii) HbVar has been linked with the GALA (Genome Alignment and Annotation database, available at http://globin.cse.psu.edu/gala/) so that users can combine information on hemoglobin variants and thalassemia mutations with a wide spectrum of genomic data. It also expands the capacity to view and analyze the data, using tools within GALA and the University of California at Santa Cruz (UCSC) Genome Browser.
Figures

References
-
- Forget B.G., Higgs,D.R., Steinberg,M. and Nagel,R.L. (2001) Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. Cambridge University Press, Cambridge, UK.
-
- Huisman T.H.J., Carver,M.F.H. and Baysal,E. (1997) A Syllabus of Thalassemia Mutations. The Sickle Cell Anemia Foundation, Augusta, GA, USA.
-
- Huisman T.H.J., Carver,M.F.H. and Efremov,G.D. (1998) A Syllabus of Human Hemoglobin Variants. 2nd edn. The Sickle Cell Anemia Foundation, Augusta, GA, USA.
-
- Hardison R.C., Chui,D.H., Giardine,B., Riemer,C., Patrinos,G.P., Anagnou,N., Miller,W. and Wajcman,H. (2002) HbVar: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Hum. Mutat., 19, 225–233. - PubMed
-
- Cao A. (2002) Carrier screening and genetic counseling in beta-thalassemia. Int. J. Hematol., 76 (Suppl. 2), 105–113. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
