

Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma
- PMID: 14685938
- PMCID: PMC1181902
- DOI: 10.1086/381054
Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma
Abstract
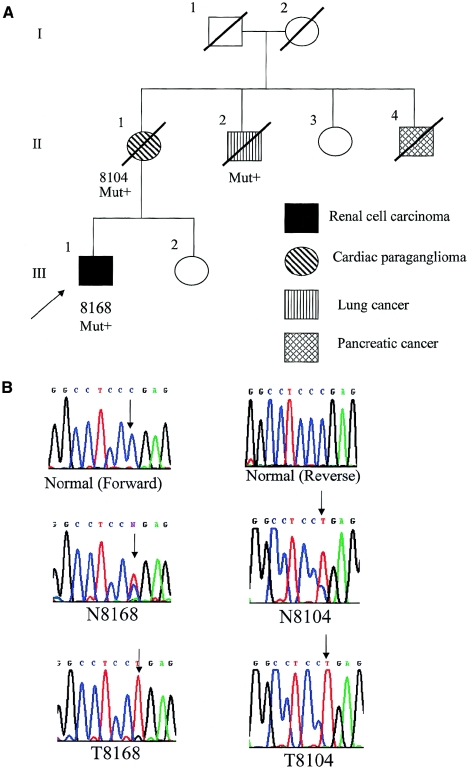
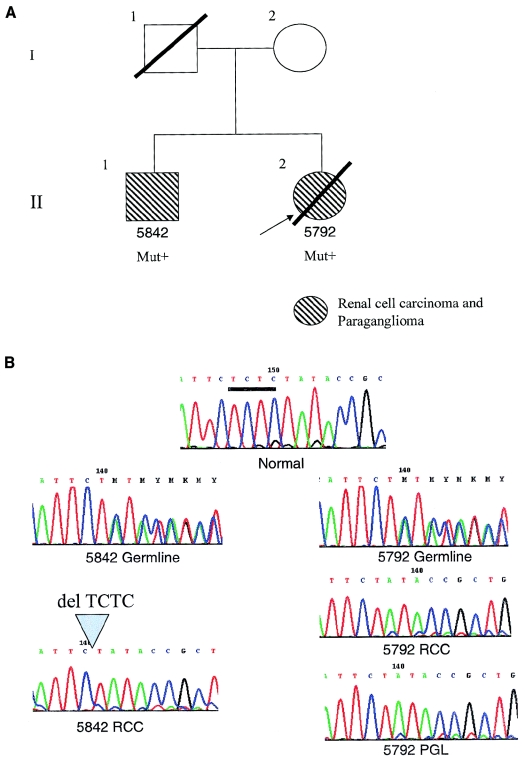
Hereditary paraganglioma syndrome has recently been shown to be caused by germline heterozygous mutations in three (SDHB, SDHC, and SDHD) of the four genes that encode mitochondrial succinate dehydrogenase. Extraparaganglial component neoplasias have never been previously documented. In a population-based registry of symptomatic presentations of phaeochromocytoma/paraganglioma comprising 352 registrants, among whom 16 unrelated registrants were SDHB mutation positive, one family with germline SDHB mutation c.847-50delTCTC had two members with renal cell carcinoma (RCC), of solid histology, at ages 24 and 26 years. Both also had paraganglioma. A registry of early-onset RCCs revealed a family comprising a son with clear-cell RCC and his mother with a cardiac tumor, both with the germline SDHB R27X mutation. The cardiac tumor proved to be a paraganglioma. All RCCs showed loss of the remaining wild-type allele. Our observations suggest that germline SDHB mutations can predispose to early-onset kidney cancers in addition to paragangliomas and carry implications for medical surveillance.
Figures
Similar articles
-
Mutation analysis of SDHB and SDHC: novel germline mutations in sporadic head and neck paraganglioma and familial paraganglioma and/or pheochromocytoma.BMC Med Genet. 2006 Jan 11;7:1. doi: 10.1186/1471-2350-7-1. BMC Med Genet. 2006. PMID: 16405730 Free PMC article.
-
Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations.JAMA. 2004 Aug 25;292(8):943-51. doi: 10.1001/jama.292.8.943. JAMA. 2004. PMID: 15328326
-
Novel succinate dehydrogenase subunit B (SDHB) mutations in familial phaeochromocytomas and paragangliomas, but an absence of somatic SDHB mutations in sporadic phaeochromocytomas.Oncogene. 2003 Mar 6;22(9):1358-64. doi: 10.1038/sj.onc.1206300. Oncogene. 2003. PMID: 12618761
-
The genetics of paragangliomas: a review.Clin Otolaryngol. 2007 Feb;32(1):7-11. doi: 10.1111/j.1365-2273.2007.01378.x. Clin Otolaryngol. 2007. PMID: 17298303 Review.
-
K40E: a novel succinate dehydrogenase (SDH)B mutation causing familial phaeochromocytoma and paraganglioma.Clin Endocrinol (Oxf). 2004 Oct;61(4):510-4. doi: 10.1111/j.1365-2265.2004.02122.x. Clin Endocrinol (Oxf). 2004. PMID: 15473885 Review.
Cited by
-
Urological cancer related to familial syndromes.Int Braz J Urol. 2017 Mar-Apr;43(2):192-201. doi: 10.1590/S1677-5538.IBJU.2016.0125. Int Braz J Urol. 2017. PMID: 27819754 Free PMC article. Review.
-
A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas.PLoS Genet. 2005 Jul;1(1):72-80. doi: 10.1371/journal.pgen.0010008. Epub 2005 Jul 25. PLoS Genet. 2005. PMID: 16103922 Free PMC article.
-
The Impact Of Succinate Dehydrogenase Gene (SDH) Mutations In Renal Cell Carcinoma (RCC): A Systematic Review.Onco Targets Ther. 2019 Sep 26;12:7929-7940. doi: 10.2147/OTT.S207460. eCollection 2019. Onco Targets Ther. 2019. PMID: 31579262 Free PMC article.
-
A study exploring critical pathways in clear cell renal cell carcinoma.Exp Ther Med. 2014 Jan;7(1):121-130. doi: 10.3892/etm.2013.1392. Epub 2013 Nov 7. Exp Ther Med. 2014. PMID: 24348776 Free PMC article.
-
Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect.Sci Rep. 2018 Feb 15;8(1):3081. doi: 10.1038/s41598-018-21361-6. Sci Rep. 2018. PMID: 29449614 Free PMC article.
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MEN 2, VHL, type 1 neurofibromatosis, SDHD, pheochromocytomas, SDHB, SDHC, SDHA, fumarase, HLRCC, FH deficiency, TSC, familial papillary RCC, hyperparathyroidism jaw tumor syndrome, and Birt-Hogg-Dubé syndrome)
References
-
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW 3rd, Cornelisse CJ, Devilee P, Devlin B (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287:848–85110.1126/science.287.5454.848 - DOI - PubMed
-
- Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH III, Myers EN, Ferrell RE, Rubinstein WS (2002) Prevalence of SDHB, SDHC and SDHD in clinic patients with head and neck paragangliomas. J Med Genet 39:178–18310.1136/jmg.39.3.178 - DOI - PMC - PubMed
-
- Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G, Green J, Hsia EY, Lamiell J, Li H, Wei MH, Schmidt L, Tory K, Kuzmin I, Stackhouse T, Latif F, Linehan M, Lerman M, Zbar B (1995) Germline mutations in the Von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat 5:66–75 - PubMed
-
- Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, Ratcliffe PJ, Maher ER (2001) Contrasting effects on HIF-1α regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet 10:1029–103810.1093/hmg/10.10.1029 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
