

Autocrine laminin-5 ligates alpha6beta4 integrin and activates RAC and NFkappaB to mediate anchorage-independent survival of mammary tumors
- PMID: 14691145
- PMCID: PMC2173718
- DOI: 10.1083/jcb.200302023
Autocrine laminin-5 ligates alpha6beta4 integrin and activates RAC and NFkappaB to mediate anchorage-independent survival of mammary tumors
Abstract
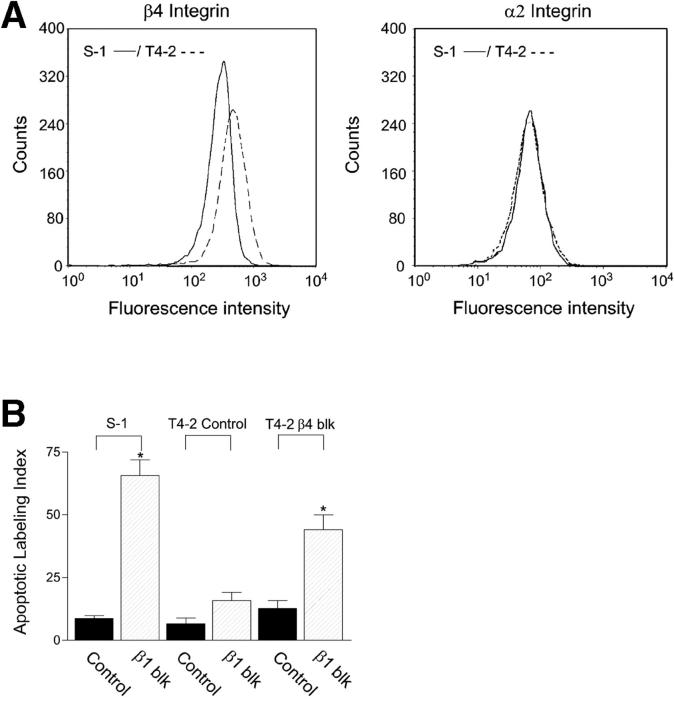
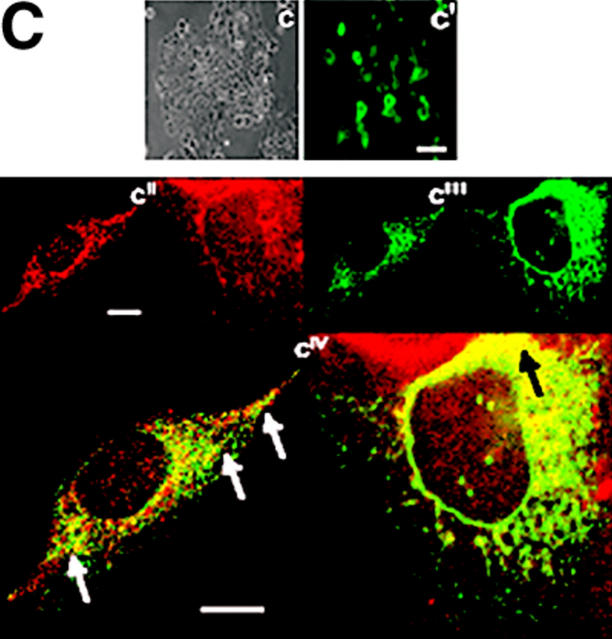
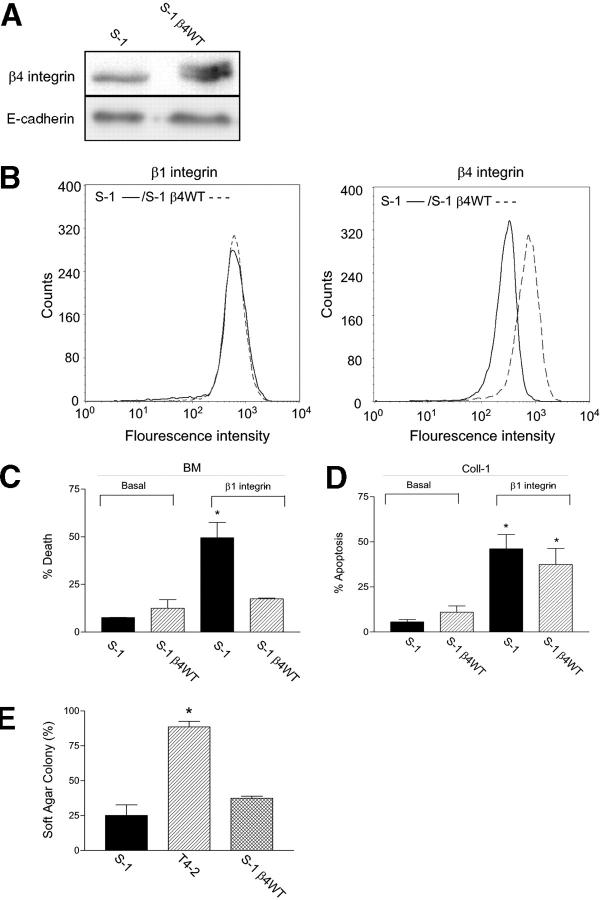
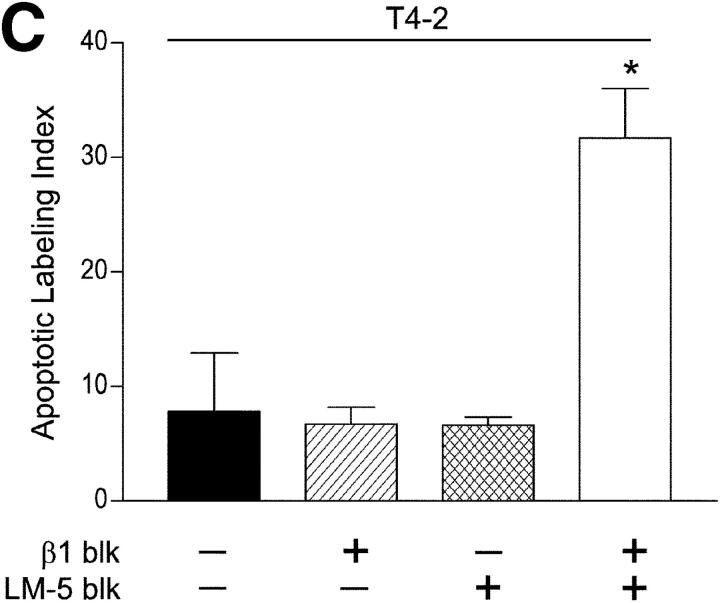
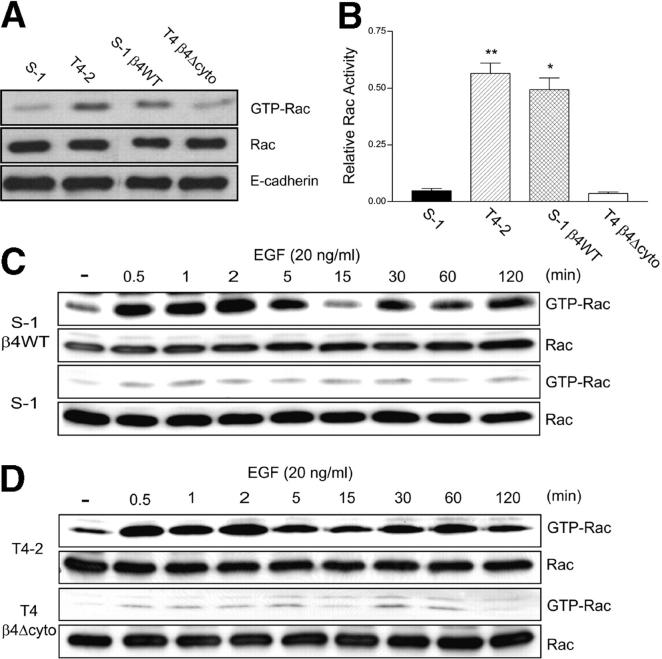
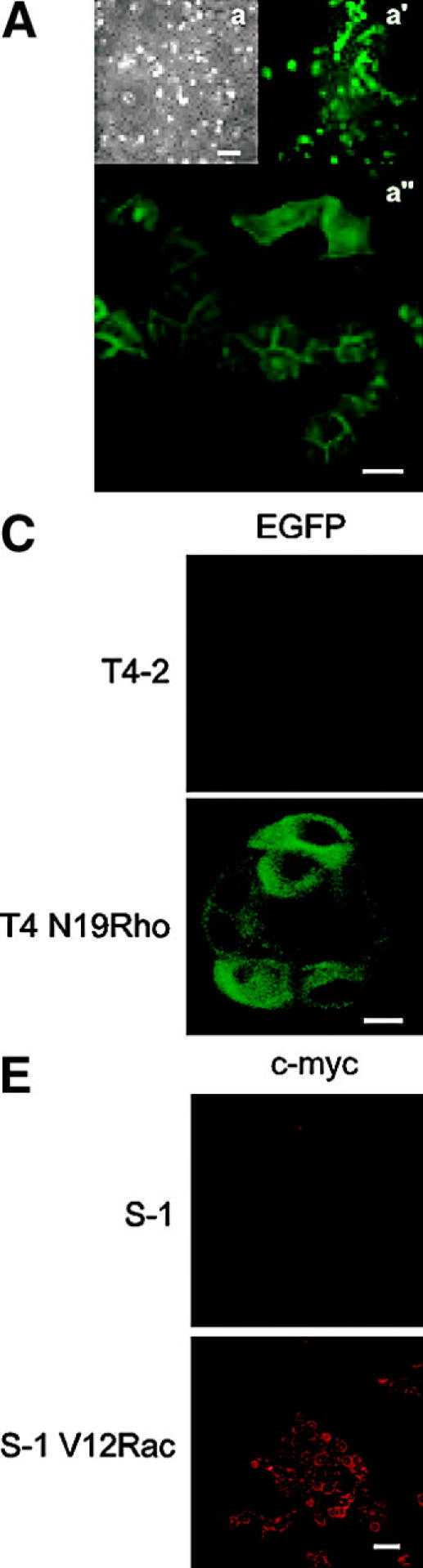
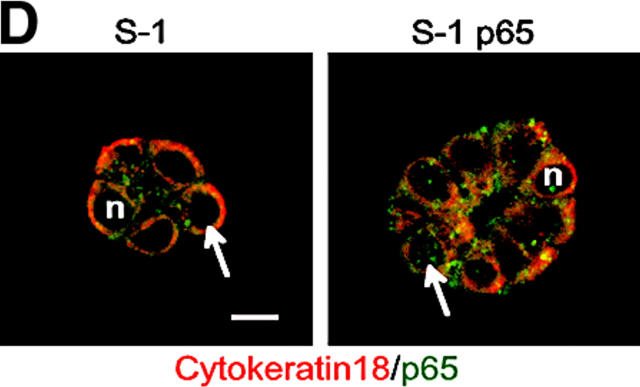
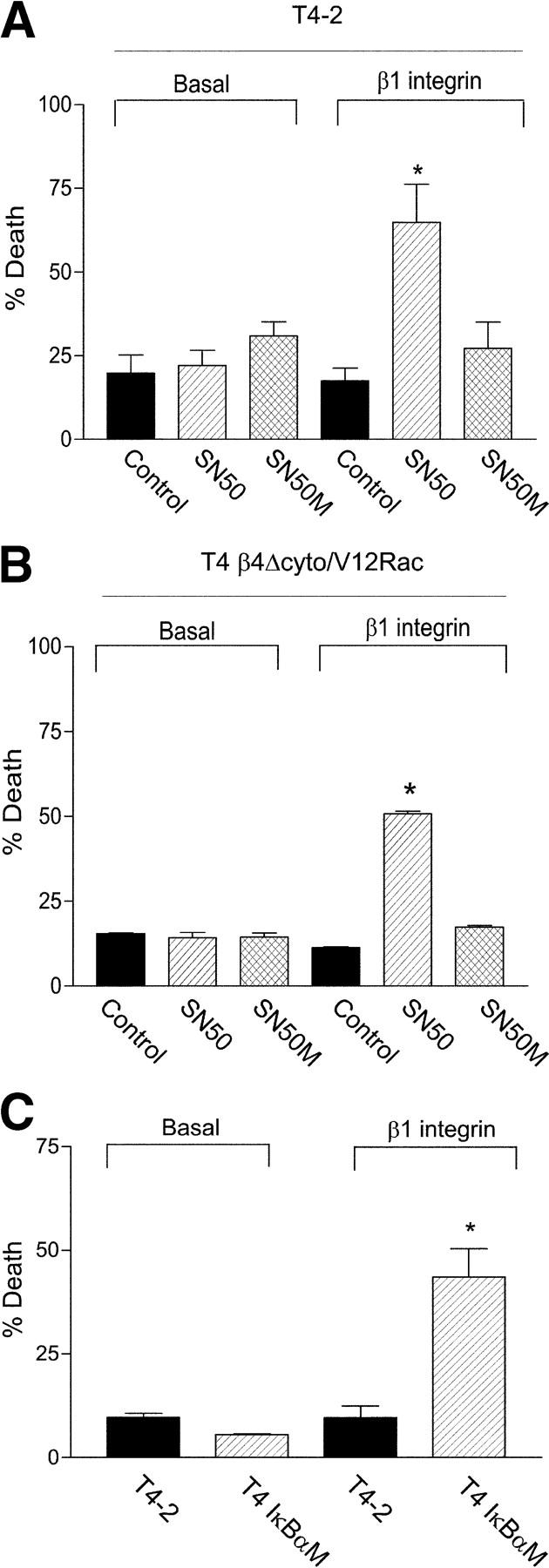
Invasive carcinomas survive and evade apoptosis despite the absence of an exogenous basement membrane. How epithelial tumors acquire anchorage independence for survival remains poorly defined. Epithelial tumors often secrete abundant amounts of the extracellular matrix protein laminin 5 (LM-5) and frequently express alpha6beta4 integrin. Here, we show that autocrine LM-5 mediates anchorage-independent survival in breast tumors through ligation of a wild-type, but not a cytoplasmic tail-truncated alpha6beta4 integrin. alpha6beta4 integrin does not mediate tumor survival through activation of ERK or AKT. Instead, the cytoplasmic tail of beta4 integrin is necessary for basal and epidermal growth factor-induced RAC activity, and RAC mediates tumor survival. Indeed, a constitutively active RAC sustains the viability of mammary tumors lacking functional beta1 and beta4 integrin through activation of NFkappaB, and overexpression of NFkappaB p65 mediates anchorage-independent survival of nonmalignant mammary epithelial cells. Therefore, epithelial tumors could survive in the absence of exogenous basement membrane through autocrine LM-5-alpha6beta4 integrin-RAC-NFkappaB signaling.
Figures













References
-
- Bozinovski, S., J.E. Jones, R. Vlahos, J.A. Hamilton, and G.P. Anderson. 2002. Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivo. J. Biol. Chem. 277:42808–42814. - PubMed
-
- Cance, W.G., J.E. Harris, M.V. Iacocca, E. Roche, X. Yang, J. Chang, S. Simkins, and L. Xu. 2000. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin. Cancer Res. 6:2417–2423. - PubMed
