

Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes
- PMID: 14694117
- PMCID: PMC368751
- DOI: 10.1128/jvi.78.2.855-867.2004
Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes
Abstract
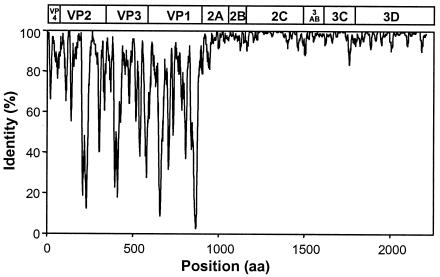
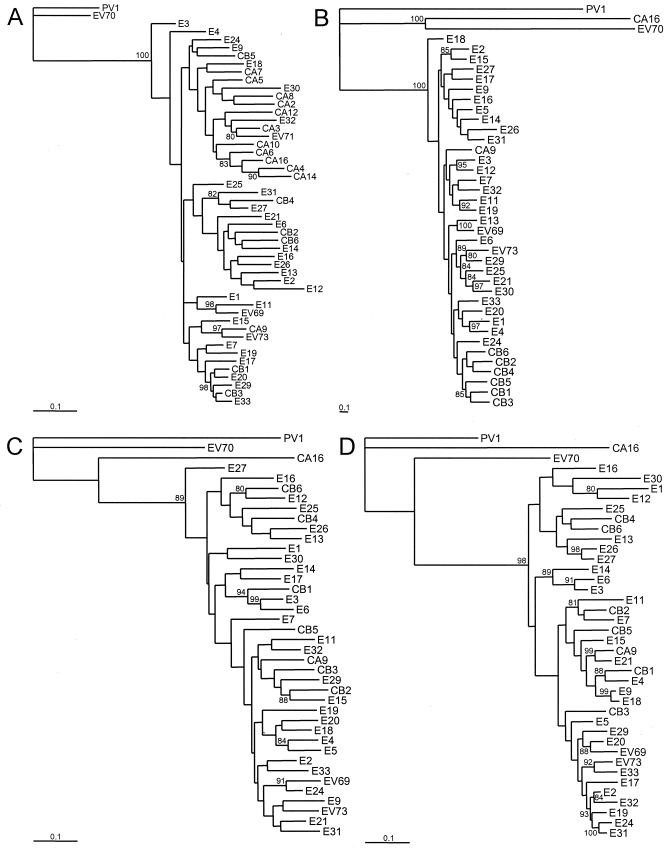
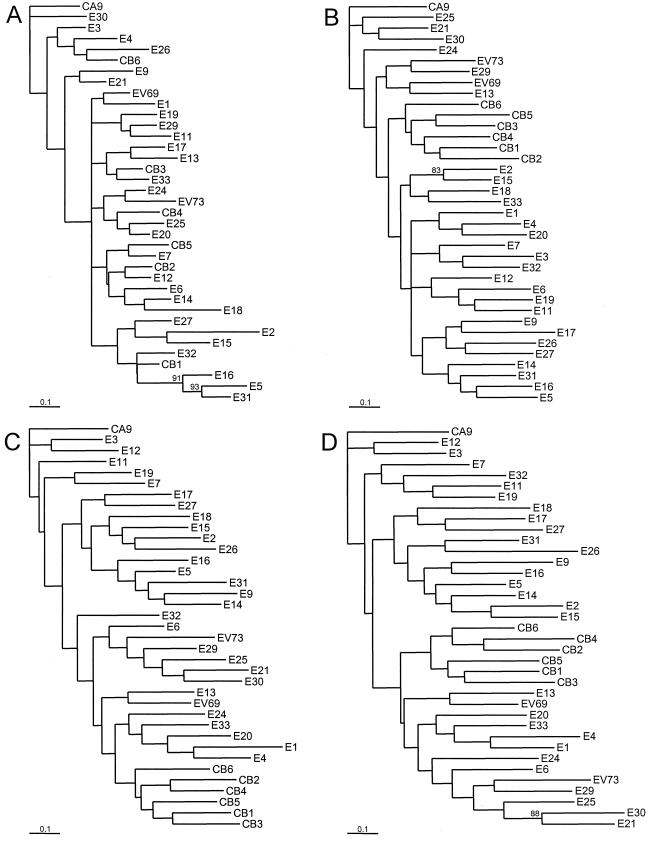
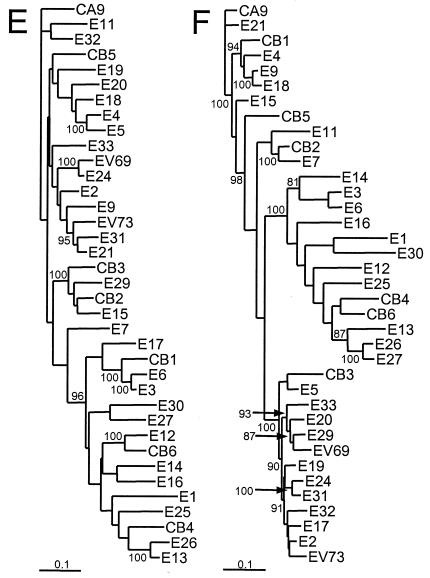
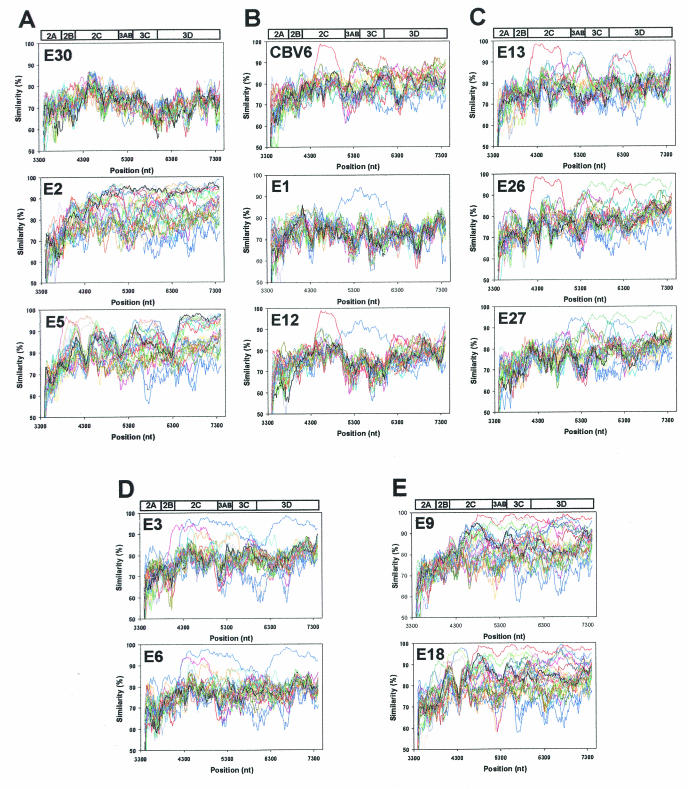
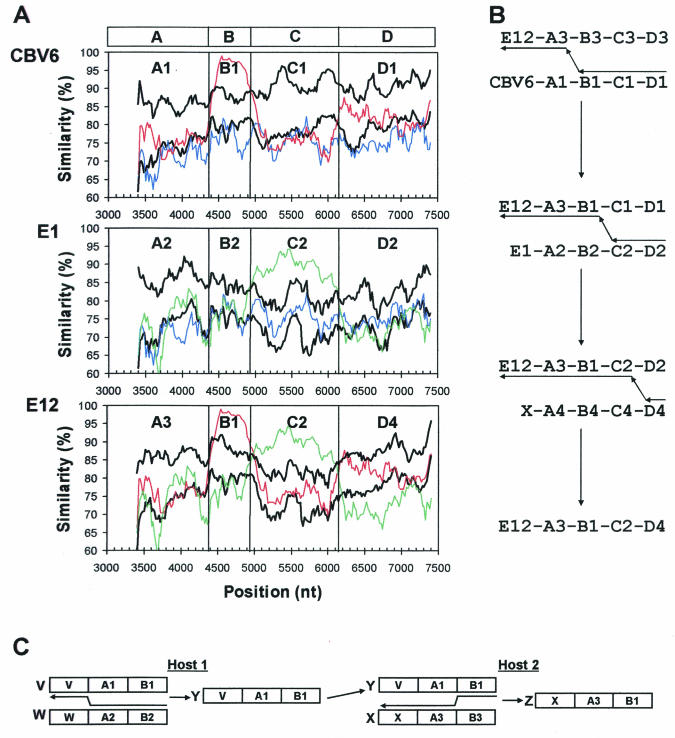
The species Human enterovirus B (HEV-B) in the family Picornaviridae consists of coxsackievirus A9; coxsackieviruses B1 to B6; echoviruses 1 to 7, 9, 11 to 21, 24 to 27, and 29 to 33; and enteroviruses 69 and 73. We have determined complete genome sequences for the remaining 22 HEV-B serotypes whose sequences were not represented in public databases and analyzed these in conjunction with previously available complete sequences in GenBank. Members of HEV-B were monophyletic relative to all other human enterovirus species in all regions of the genome except in the 5'-nontranslated region (NTR), where they are known to cluster with members of HEV-A. Within HEV-B, phylogenies constructed from the structural (P1) and nonstructural regions of the genome (P2 and P3) are incongruent, suggesting that recombination had occurred. Similarity plots and bootscanning analysis across the complete genome identified multiple sites at which the phylogeny of a given strain's sequence shifted, indicating potential recombination points. These points are distributed in the 5'-NTR and throughout P2 and P3, but no sites with >80% bootstrap support were identified within the capsid. Individual sequence comparisons and phylogenetic analyses suggest that members of HEV-B have recombined with one another on multiple occasions, resulting in a complex mosaic of sequences derived from multiple parental viruses in the nonstructural regions of the genome. We conclude that RNA recombination is a common mechanism for enterovirus evolution and that recombination within the nonstructural regions of the genome (P2 and P3) has been observed only among members of the same species.
Figures
References
-
- Andersson, P., K. Edman, and A. M. Lindberg. 2002. Molecular analysis of the echovirus 18 prototype: evidence of interserotypic recombination with echovirus 9. Virus Res. 85:71-83. - PubMed
-
- Blomqvist, S., A.-L. Bruu, M. Stenvik, and T. Hovi. 2003. Characterization of a recombinant type 3/type 2 poliovirus isolated from a healthy vaccinee and containing a chimeric capsid protein VP1. J. Gen. Virol. 84:573-580. - PubMed
-
- Branche, W. C., V. M. Young, F. M. Houston, and L. W. Koontz. 1965. Characterization of prototype virus ECHO-32. Proc. Soc. Exp. Biol. Med. 118:186-190. - PubMed
-
- Cammack, N., A. Phillips, G. Dunn, V. Patel, and P. D. Minor. 1988. Intertypic genomic rearrangements of poliovirus strains in vaccinees. Virology 167:507-514. - PubMed
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
