

Reversing established sepsis with antagonists of endogenous high-mobility group box 1
- PMID: 14695889
- PMCID: PMC314179
- DOI: 10.1073/pnas.2434651100
Reversing established sepsis with antagonists of endogenous high-mobility group box 1
Abstract
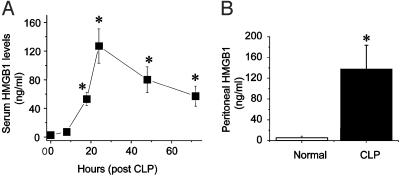
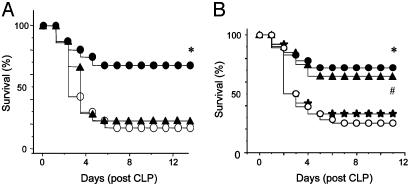
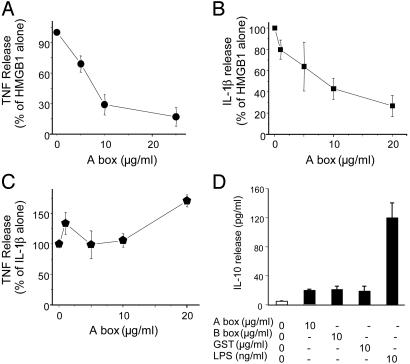
Despite significant advances in intensive care therapy and antibiotics, severe sepsis accounts for 9% of all deaths in the United States annually. The pathological sequelae of sepsis are characterized by a systemic inflammatory response, but experimental therapeutics that target specific early inflammatory mediators [tumor necrosis factor (TNF) and IL-1beta] have not proven efficacious in the clinic. We recently identified high mobility group box 1 (HMGB1) as a late mediator of endotoxin-induced lethality that exhibits significantly delayed kinetics relative to TNF and IL-1beta. Here, we report that serum HMGB1 levels are increased significantly in a standardized model of murine sepsis, beginning 18 h after surgical induction of peritonitis. Specific inhibition of HMGB1 activity [with either anti-HMGB1 antibody (600 microg per mouse) or the DNA-binding A box (600 microg per mouse)] beginning as late as 24 h after surgical induction of peritonitis significantly increased survival (nonimmune IgG-treated controls = 28% vs. anti-HMGB1 antibody group = 72%, P < 0.03; GST control protein = 28% vs. A box = 68%, P < 0.03). Animals treated with either HMGB1 antagonist were protected against the development of organ injury, as evidenced by improved levels of serum creatinine and blood urea nitrogen. These observations demonstrate that specific inhibition of endogenous HMGB1 therapeutically reverses lethality of established sepsis indicating that HMGB1 inhibitors can be administered in a clinically relevant time frame.
Figures



References
-
- Munford, R. S. & Pugin, J. (2001) Am. J. Respir. Crit. Care Med. 163, 316-321. - PubMed
-
- Angus, D. & Wax, R. S. (2001) Crit. Care Med. 29, Suppl. 7, S109-S116. - PubMed
-
- Friedman, G., Silva, E. & Vincent, J. L. (1998) Crit. Care Med. 26, 2078-2086. - PubMed
-
- Tracey, K. J., Beutler, B., Lowry, S. F., Merryweather, J., Wolpe, S., Milsark, I. W., Hariri, R. J., Fahey, T. J., III, Zentelia, A., Albert, J. D., et al. (1986) Science 234, 470-474. - PubMed
-
- Tracey, K. J., Fong, Y., Hesse, D. G., Manogue, K. R., Lee, A. T., Kuo, G. C., Lowry, S. F. & Cerami, A. (1987) Nature 330, 662-664. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
