

BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha
- PMID: 14699081
- PMCID: PMC1887730
- DOI: 10.1084/jem.20030613
BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha
Abstract
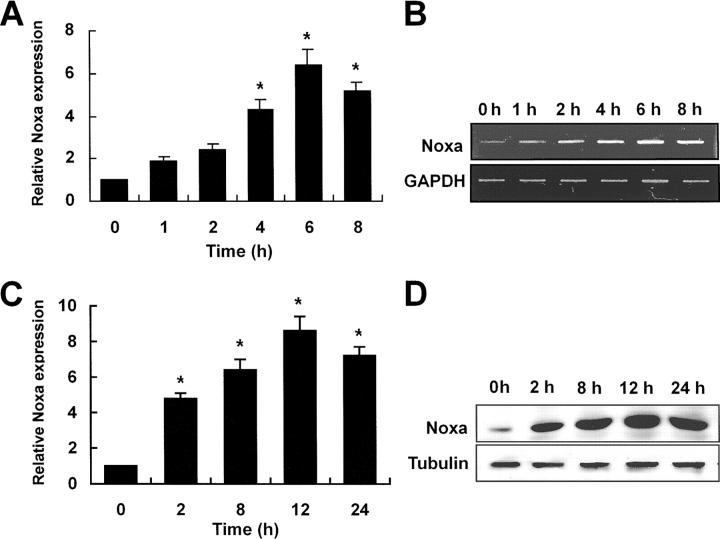
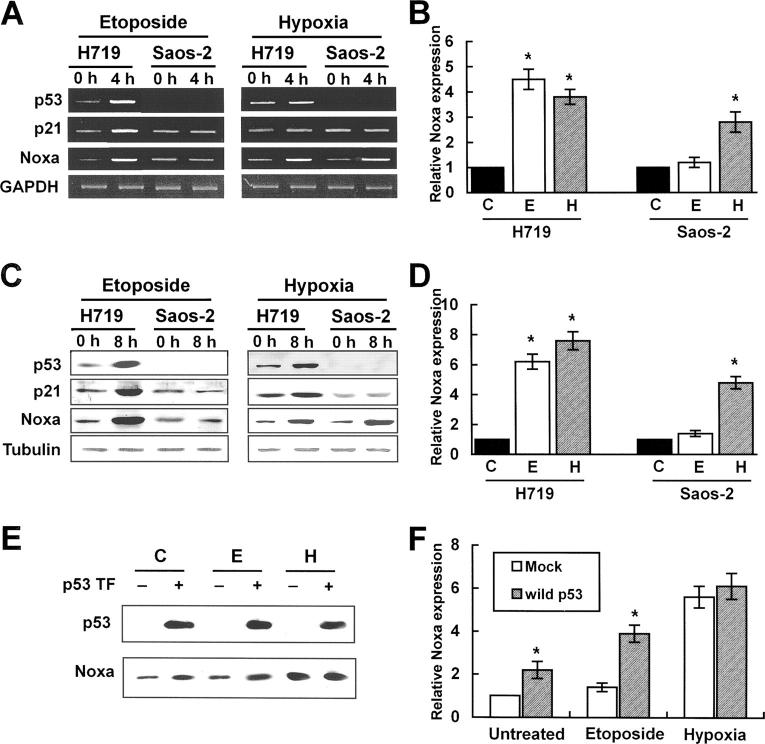
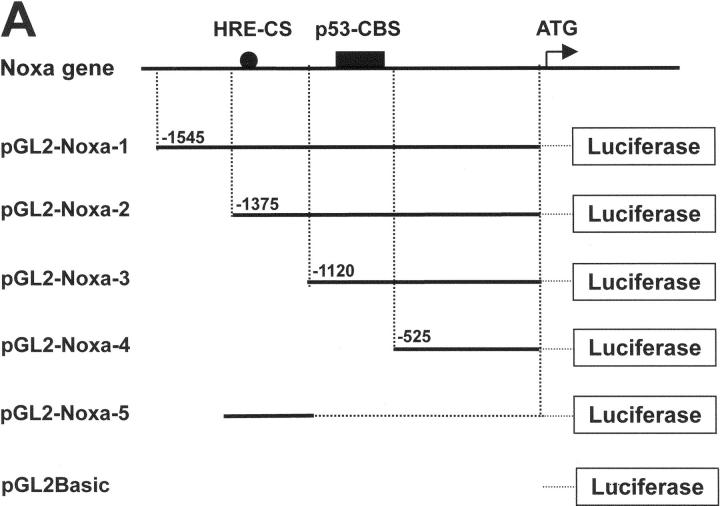
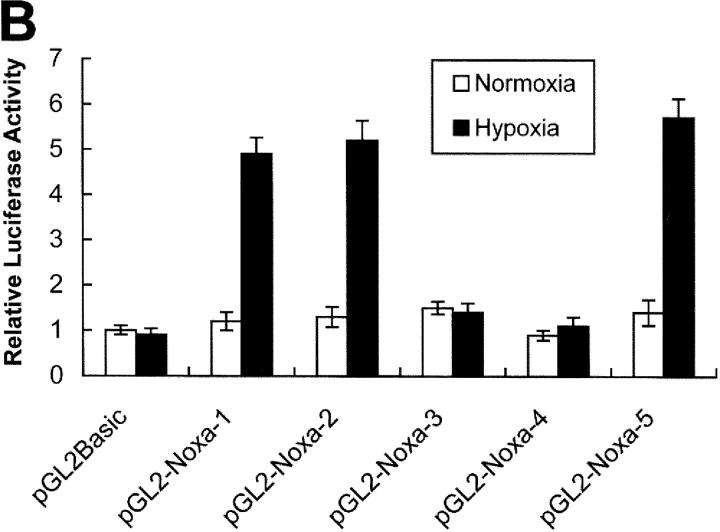
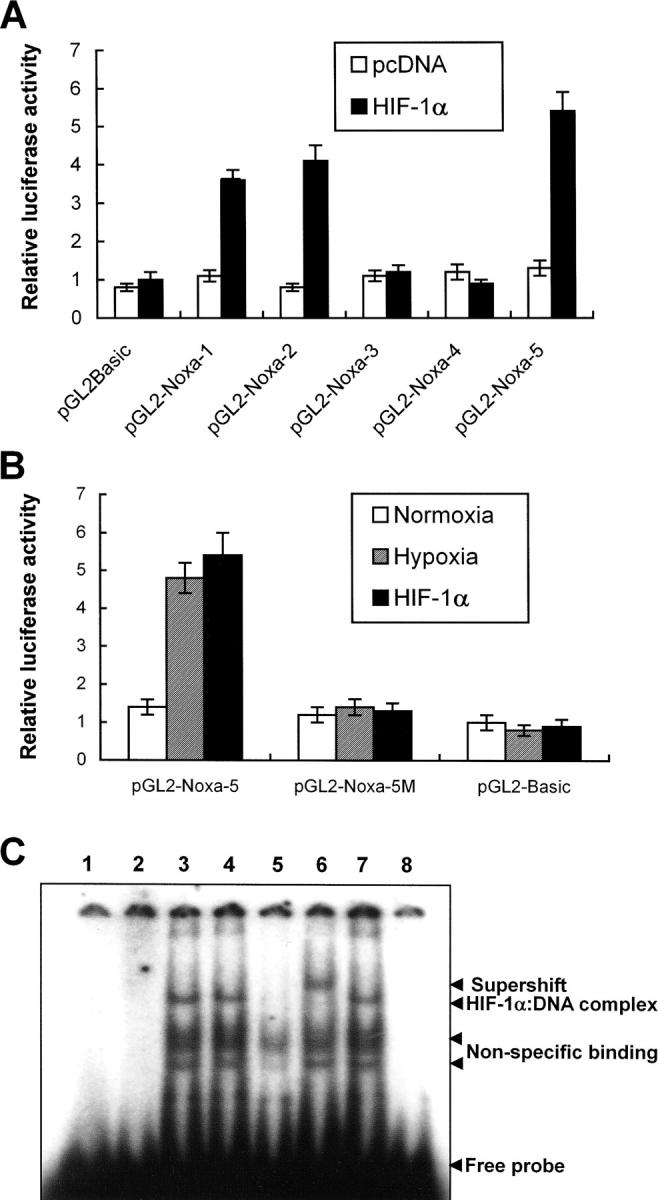
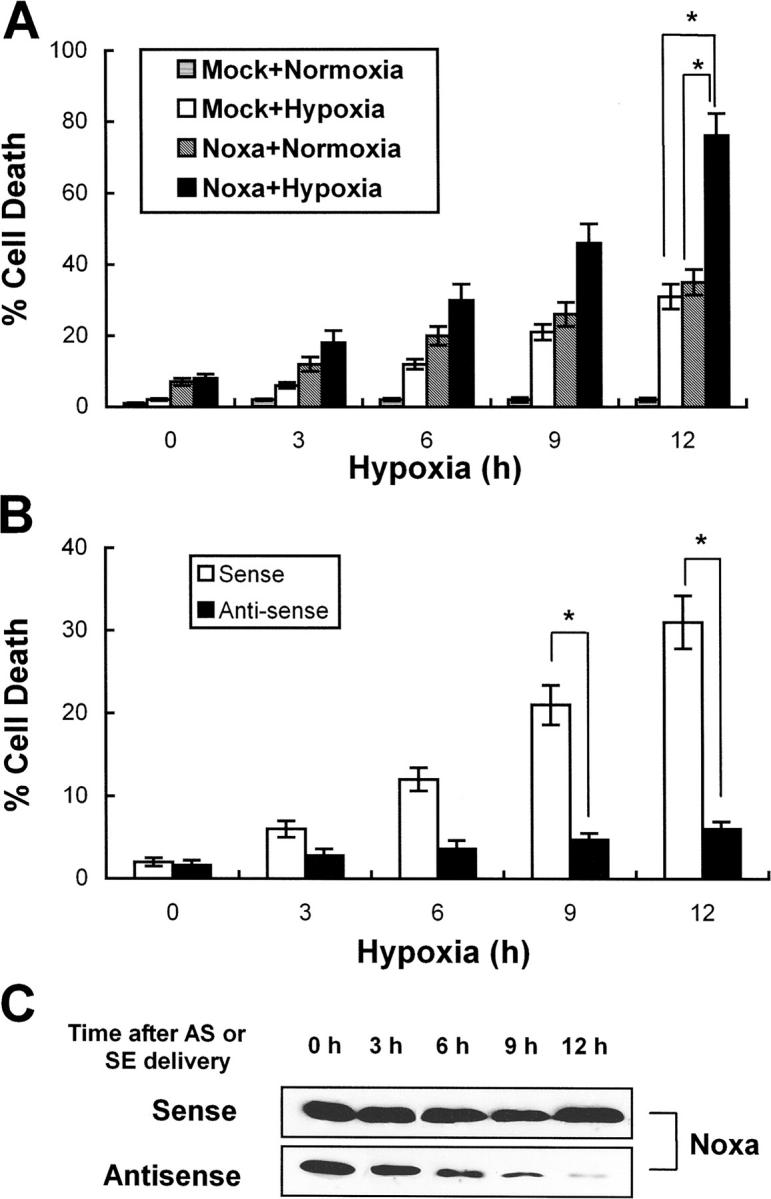
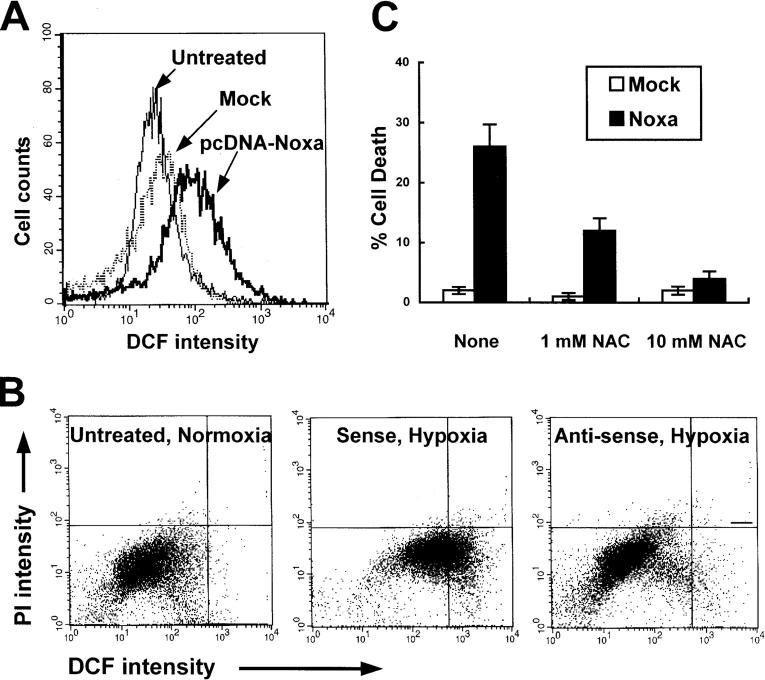
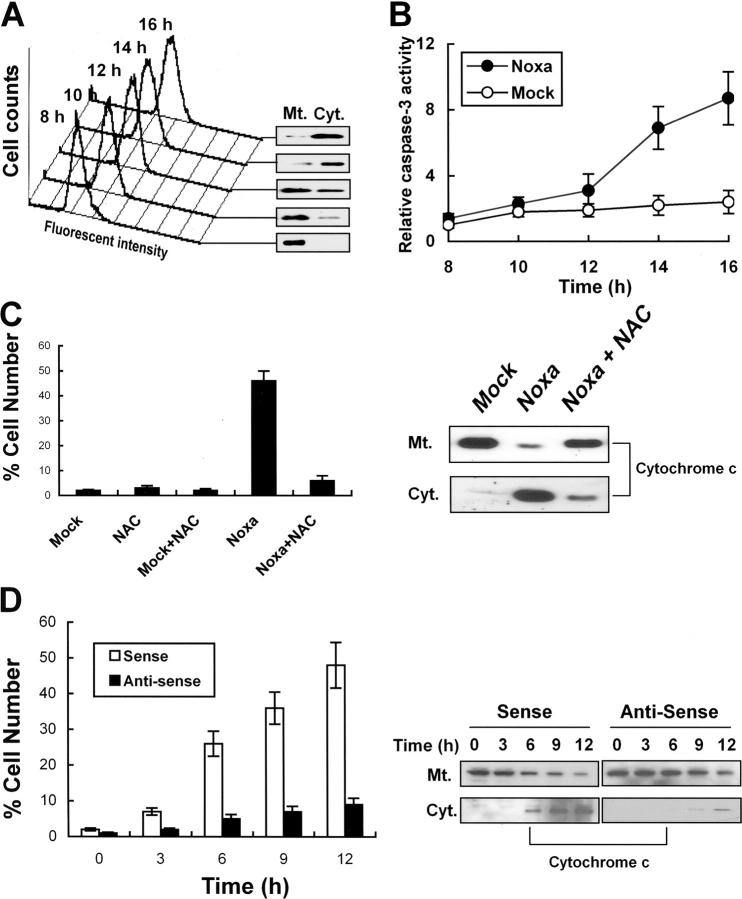
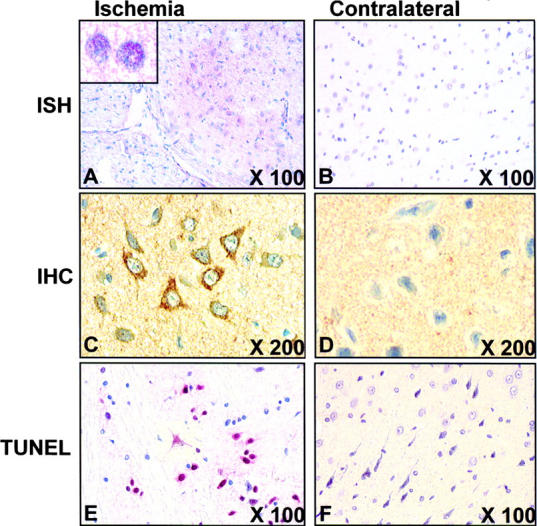
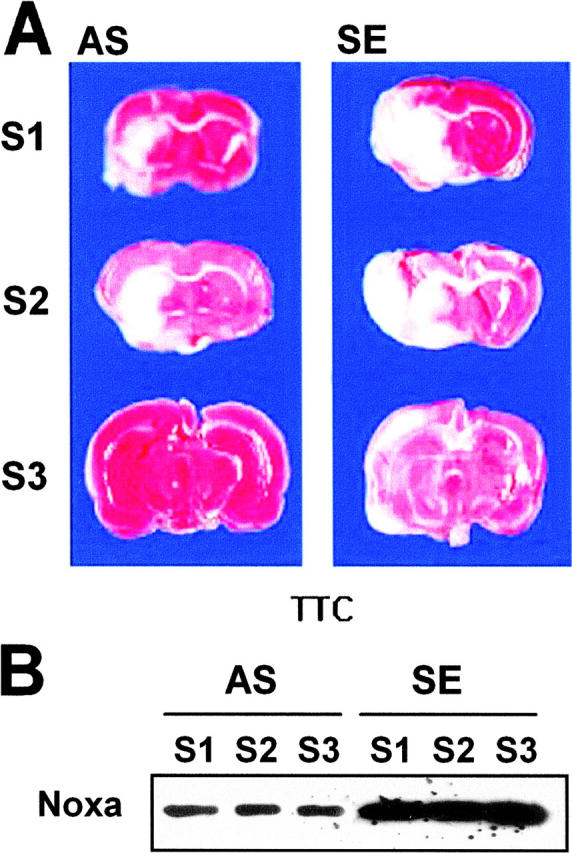
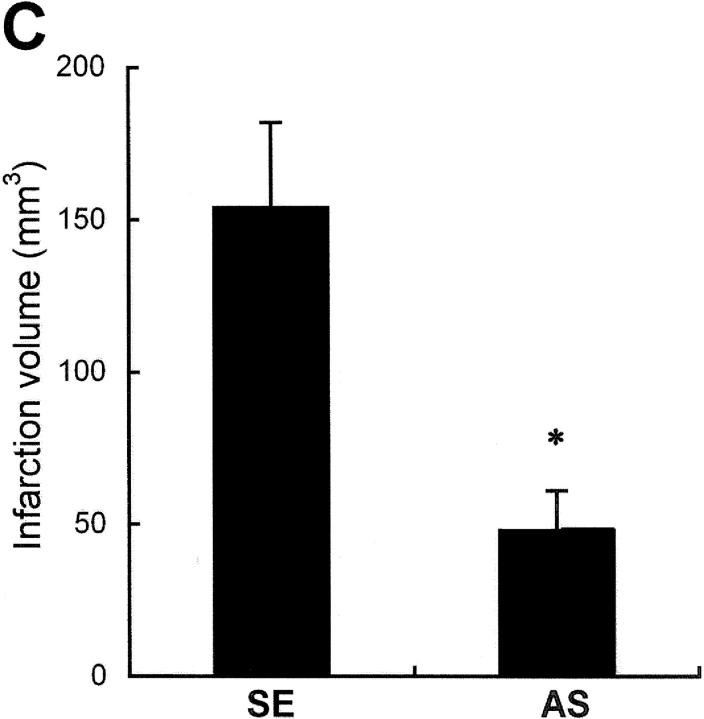
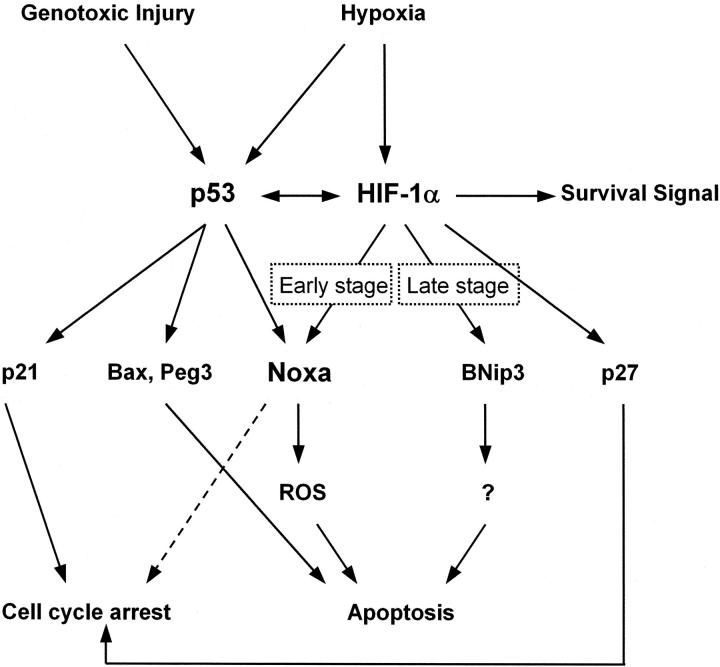
Hypoxia is a common cause of cell death and is implicated in many disease processes including stroke and chronic degenerative disorders. In response to hypoxia, cells express a variety of genes, which allow adaptation to altered metabolic demands, decreased oxygen demands, and the removal of irreversibly damaged cells. Using polymerase chain reaction-based suppression subtractive hybridization to find genes that are differentially expressed in hypoxia, we identified the BH3-only Bcl-2 family protein Noxa. Noxa is a candidate molecule mediating p53-induced apoptosis. We show that Noxa promoter responds directly to hypoxia via hypoxia-inducible factor (HIF)-1alpha. Suppression of Noxa expression by antisense oligonucleotides rescued cells from hypoxia-induced cell death and decreased infarction volumes in an animal model of ischemia. Further, we show that reactive oxygen species and resultant cytochrome c release participate in Noxa-mediated hypoxic cell death. Altogether, our results show that Noxa is induced by HIF-1alpha and mediates hypoxic cell death.
Figures
References
-
- Halterman, M.W., and H.J. Federoff. 1999. HIF-1α and p53 promote hypoxia-induced delayed neuronal death in models of CNS ischemia. Exp. Neurol. 159:65–72. - PubMed
-
- Bunn, H.F., and R.O. Poyton. 1996. Oxygen sensing and molecular adaptation to hypoxia. Physiol. Rev. 76:839–885. - PubMed
-
- Semenza, G.L. 1999. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15:551–578. - PubMed
-
- Gardner, L.B., Q. Li, M.S. Park, W.M. Flanagan, G.L. Semenza, and C.V. Dang. 2001. Hypoxia inhibits G1/S transition through regulation of p27 expression. J. Biol. Chem. 276:7919–7926. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
