

A permissive retinoid X receptor/thyroid hormone receptor heterodimer allows stimulation of prolactin gene transcription by thyroid hormone and 9-cis-retinoic acid
- PMID: 14701725
- PMCID: PMC343792
- DOI: 10.1128/MCB.24.2.502-513.2004
A permissive retinoid X receptor/thyroid hormone receptor heterodimer allows stimulation of prolactin gene transcription by thyroid hormone and 9-cis-retinoic acid
Abstract
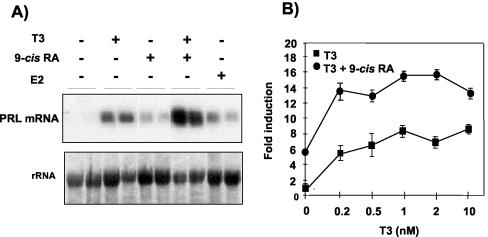
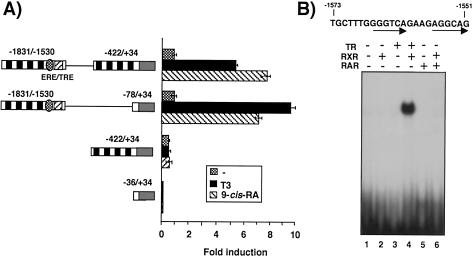
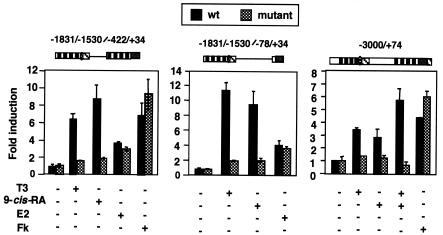
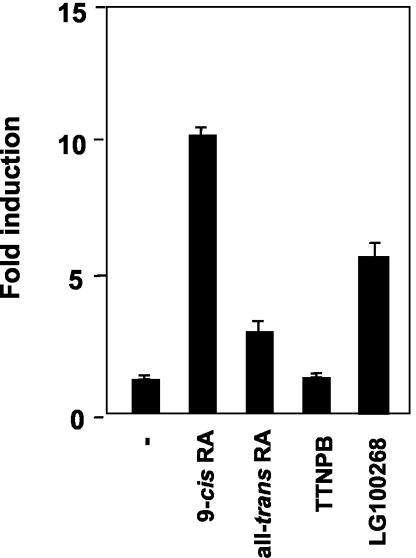
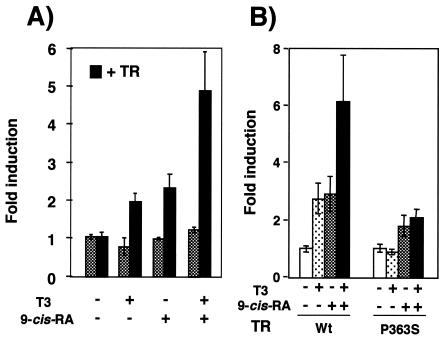
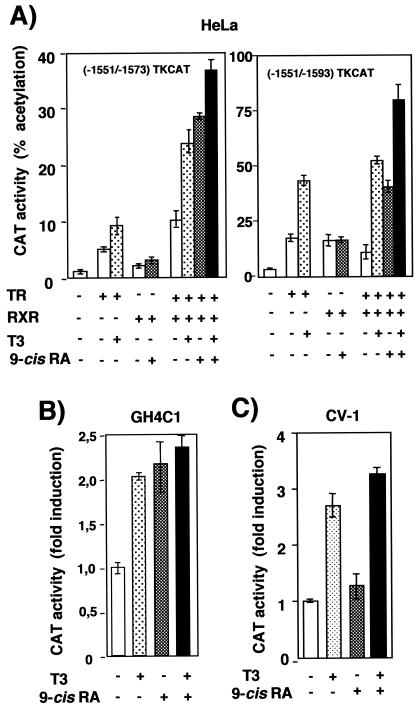
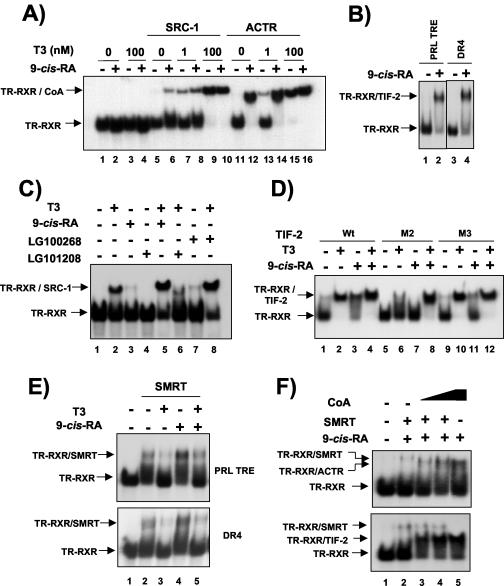
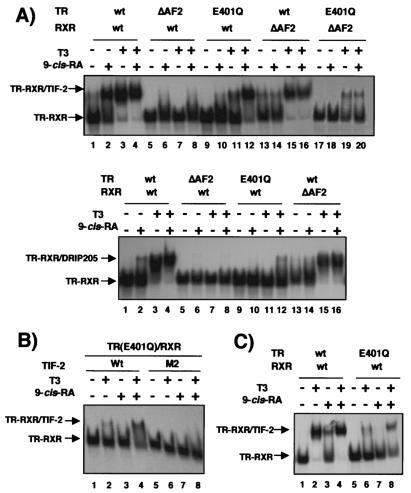
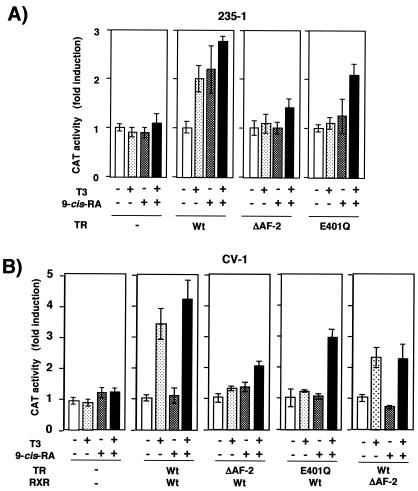
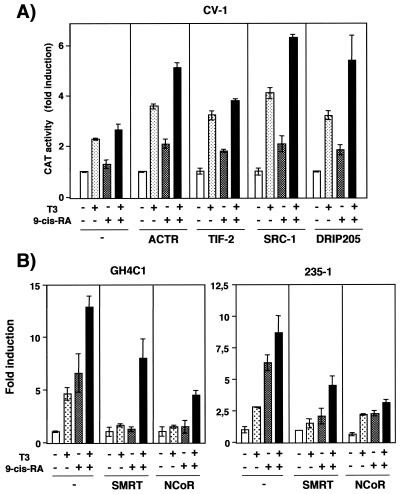
Heterodimers of the retinoid X receptor (RXR) with the thyroid hormone receptor (TR) are considered to be nonpermissive. It is believed that within these complexes RXR acts as a "silent partner." We demonstrate here that a permissive heterodimer mediates stimulation of prolactin expression by the thyroid hormone T3 and by 9-cis retinoic acid (9-cis-RA). A response element located in the prolactin distal enhancer mediates transactivation by both ligands in pituitary cells, and RXR recruits coactivators when bound to this element as a heterodimer with TR. Furthermore, transcription by the RXR agonist can be obtained in CV-1 cells only after overexpression of coactivators, and overexpression of corepressors inhibits the response in pituitary cells. Thus, cell type-specific differences in coregulator recruitment can determine the cellular response to both ligands. Coactivator recruitment by 9-cis-RA requires the ligand-dependent transactivation domains (AF-2) of both heterodimeric partners. Interestingly, the presence of the RXR ligand can overcome the deleterious effect of the AF-2 mutation E401Q on association with coactivators and transactivation. These results demonstrate an unexpected role for RXR in TR signaling and show that in particular cellular environments this receptor can act as a "nonsilent" partner of TR, allowing stimulation by RXR agonists.
Figures
References
-
- Aranda, A., and A. Pascual. 2001. Nuclear hormone receptors and gene expression. Physiol. Rev. 81:1269-1304. - PubMed
-
- Bedó, G., P. Santisteban, and A. Aranda. 1989. Retinoic acid regulates growth hormone gene expression. Nature 339:231-234. - PubMed
-
- Castillo, A. I., A. M. Jimenez-Lara, R. M. Tolon, and A. Aranda. 1999. Synergistic activation of the prolactin promoter by vitamin D receptor and GHF-1: role of the coactivators CREB-binding protein and steroid hormone receptor coactivator 1. Mol. Endocrinol. 13:1141-1154. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources