

Dopamine D1/D5 receptor modulates state-dependent switching of soma-dendritic Ca2+ potentials via differential protein kinase A and C activation in rat prefrontal cortical neurons
- PMID: 14715933
- PMCID: PMC6729575
- DOI: 10.1523/JNEUROSCI.1650-03.2004
Dopamine D1/D5 receptor modulates state-dependent switching of soma-dendritic Ca2+ potentials via differential protein kinase A and C activation in rat prefrontal cortical neurons
Abstract
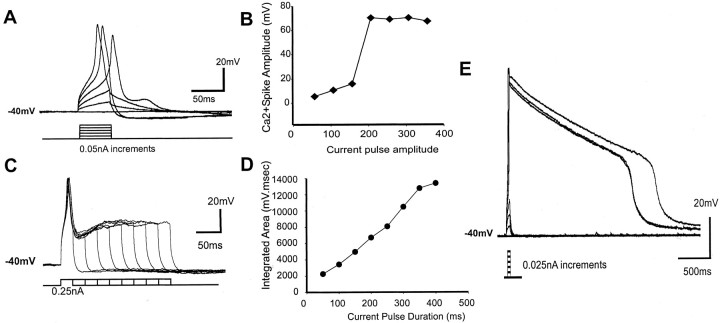
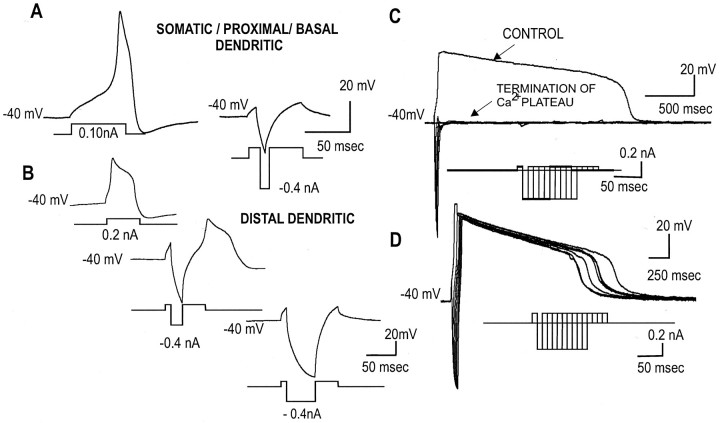
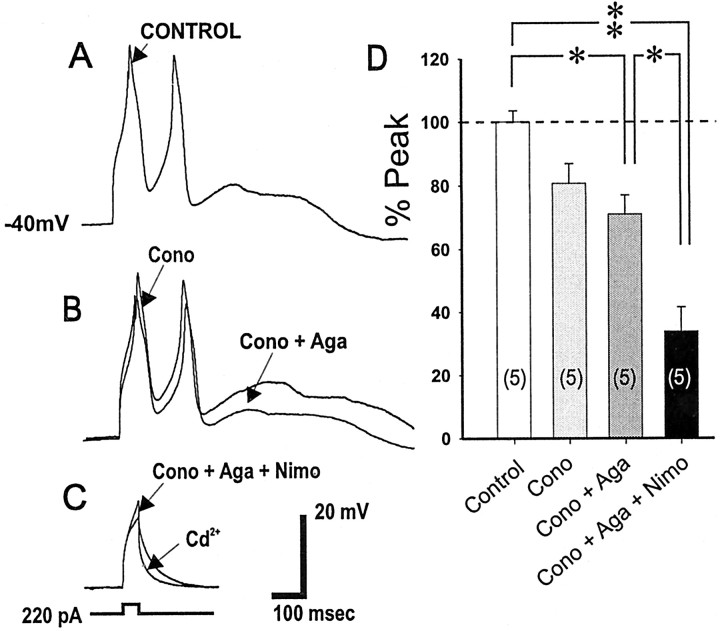
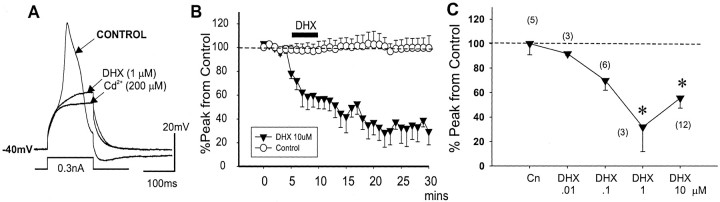
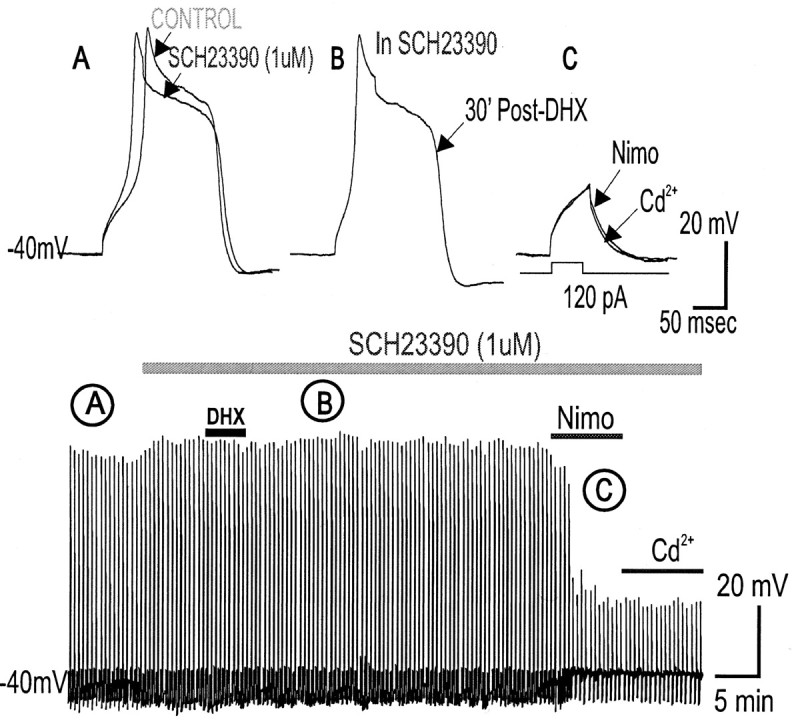
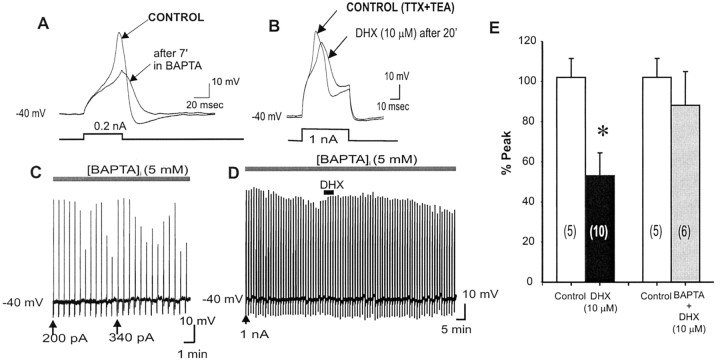
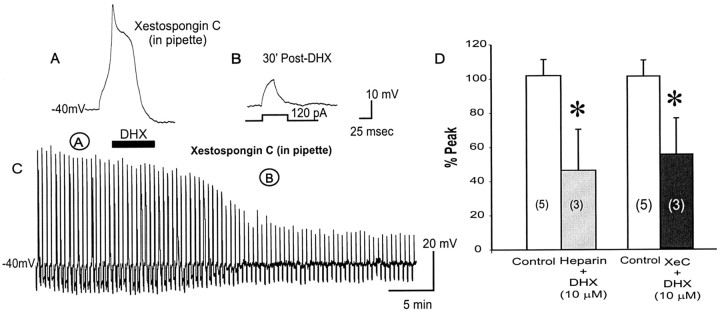
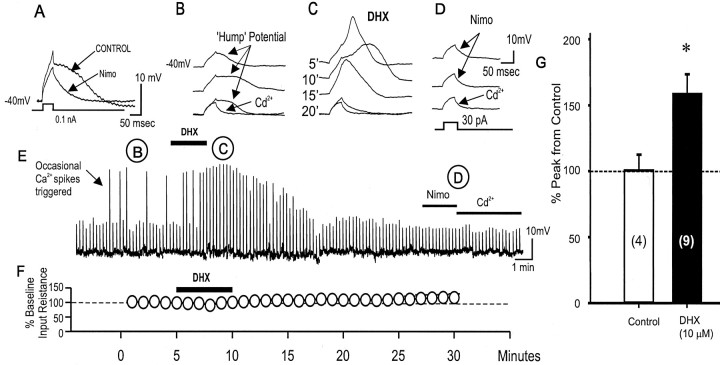
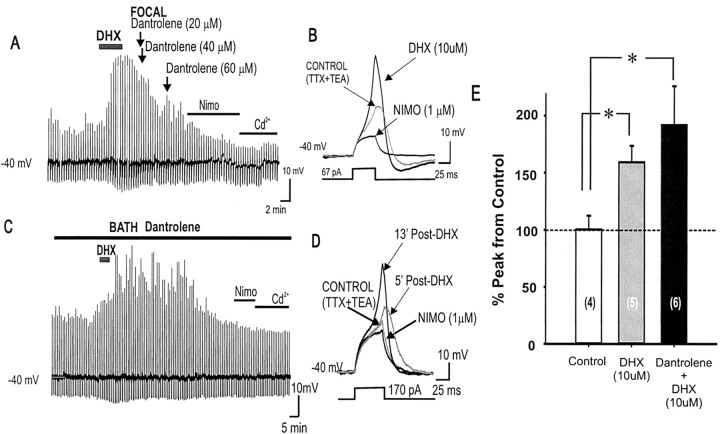
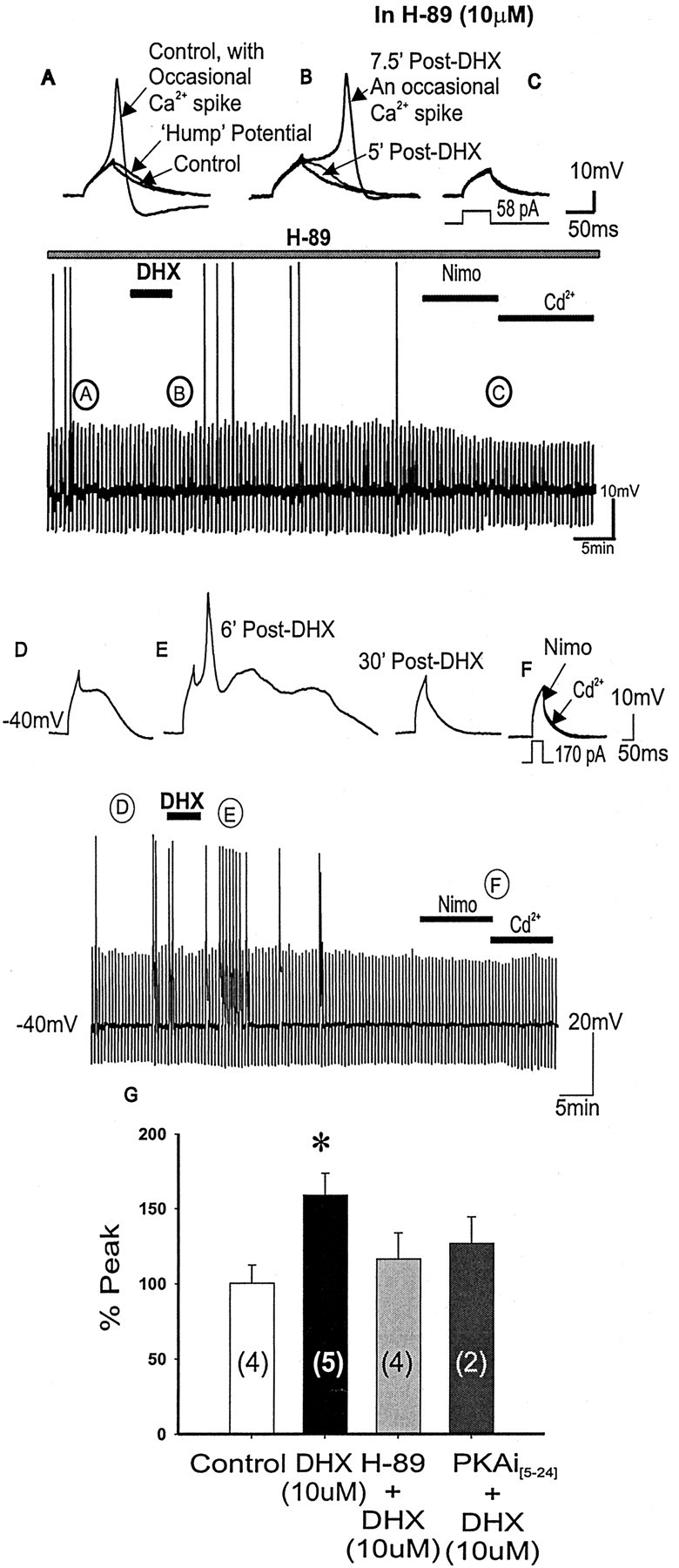
To determine the nature of dopamine modulation of dendritic Ca2+ signaling in layers V-VI prefrontal cortex (PFC) neurons, whole-cell Ca2+ potentials were evoked after blockade of Na+ and K+ channels. Soma-dendritic Ca2+ spikes evoked by suprathreshold depolarizing pulses, which could be terminated by superimposed brief intrasomatic hyperpolarizing pulses, are blocked by the L-type Ca2+ channel antagonist nimodipine (1 microM). The D1/D5 receptor agonist dihydrexidine (DHX) (0.01-10 microM; 5 min) or R-(+)SKF81291 (10 microM) induced a prolonged (>30 min) dose-dependent peak suppression of these Ca2+ spikes. This effect was dependent on [Ca2+]i- and protein kinase C (PKC)-dependent mechanisms because [Ca2+]i chelation by BAPTA or inhibition of PKC by bisindolymaleimide (BiM1), but not inhibition of [Ca2+]i release with heparin or Xestospongin C, prevented the D1-mediated suppression of Ca2+ spikes. Depolarizing pulses subthreshold to activating a Ca2+ spike evoked a nimodipine-sensitive Ca2+ "hump" potential. D1/D5 stimulation induced an N-[2-((o-bromocinamyl)amino)ethyl]-5-isoquinolinesulfonamide (H-89)- or internal PKA inhibitory peptide[5-24]-sensitive (PKA-dependent) transient (approximately 7 min) potentiation of the hump potential to full Ca2+ spike firing. Furthermore, application of DHX in the presence of the PKC inhibitor BiM1 or internal PKC inhibitory peptide[19-36] resulted in persistent firing of full Ca2+ spike bursts, suggesting that a D1/D5-PKA mechanism switches subthreshold Ca2+ hump potential to fire full Ca2+ spikes, which are eventually turned off by a D1/D5-Ca2+-dependent PKC mechanism. This depolarizing state-dependent, D1/D5-activated, bi-directional switching of soma-dendritic L-type Ca2+ channels via PKA-dependent potentiation and PKC-dependent suppression may provide spatiotemporal regulation of synaptic integration and plasticity in PFC.
Figures




References
-
- Artalejo CR, Ariano MA, Perlman RL, Fox AP (1990) Activation of facilitation calcium channels in chromaffin cells by D1 dopamine receptors through a cAMP/protein kinase A-dependent mechanism. Nature 348: 239-242. - PubMed
-
- Artalejo CR, Rossie S, Perlman RL, Fox AP (1992) Voltage-dependent phosphorylation may recruit Ca2+ current facilitation in chromaffin cells. Nature 358: 63-66. - PubMed
-
- Berger B, Gaspar P, Verney C (1991) Dopaminergic innervation of the cerebral cortex: unexpected differences between rodents and primates. Trends Neurosci 14: 21-27. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous