

The folding landscape of Streptomyces griseus protease B reveals the energetic costs and benefits associated with evolving kinetic stability
- PMID: 14718653
- PMCID: PMC2286692
- DOI: 10.1110/ps.03336804
The folding landscape of Streptomyces griseus protease B reveals the energetic costs and benefits associated with evolving kinetic stability
Abstract
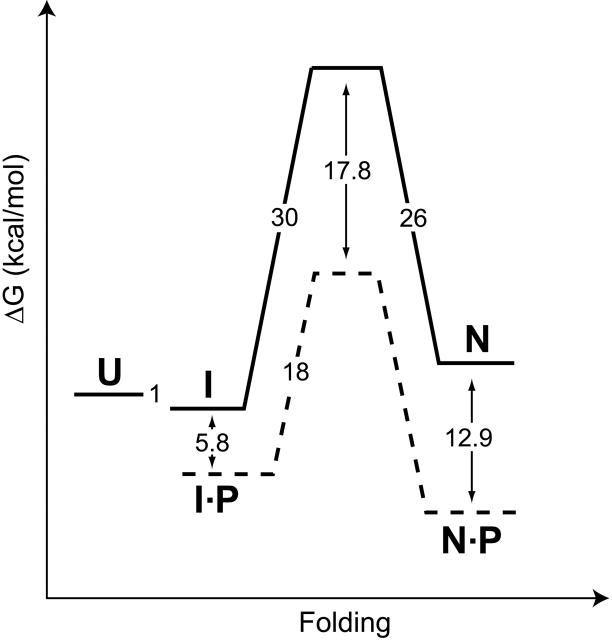
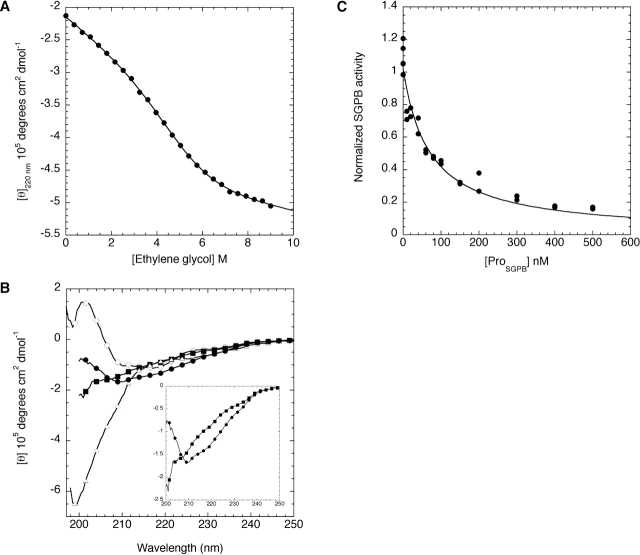
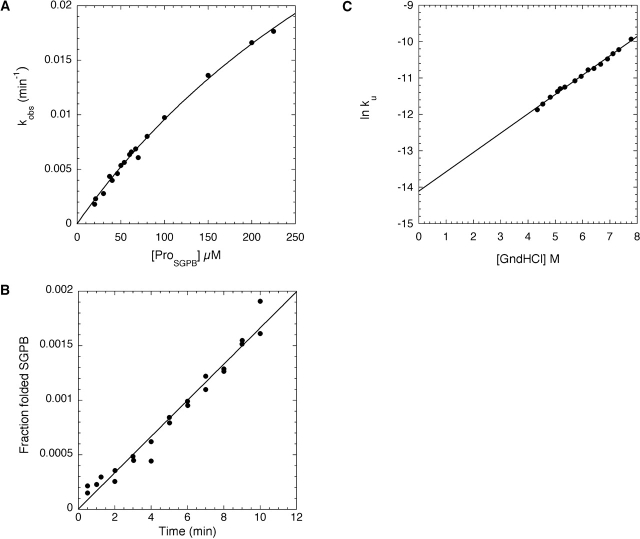
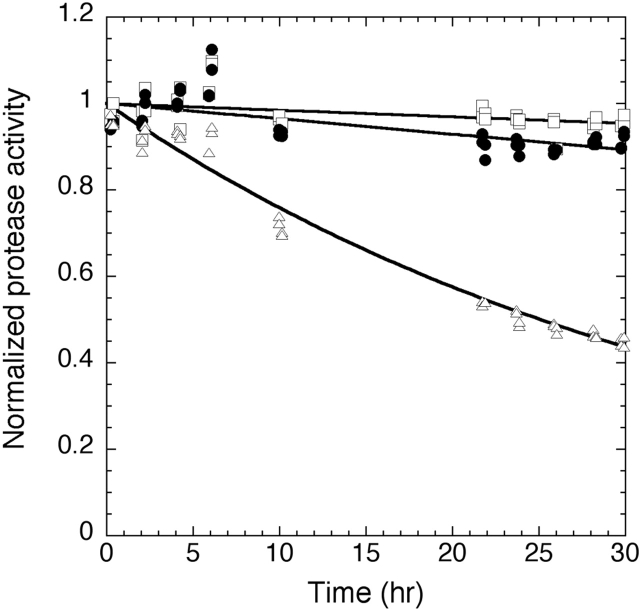
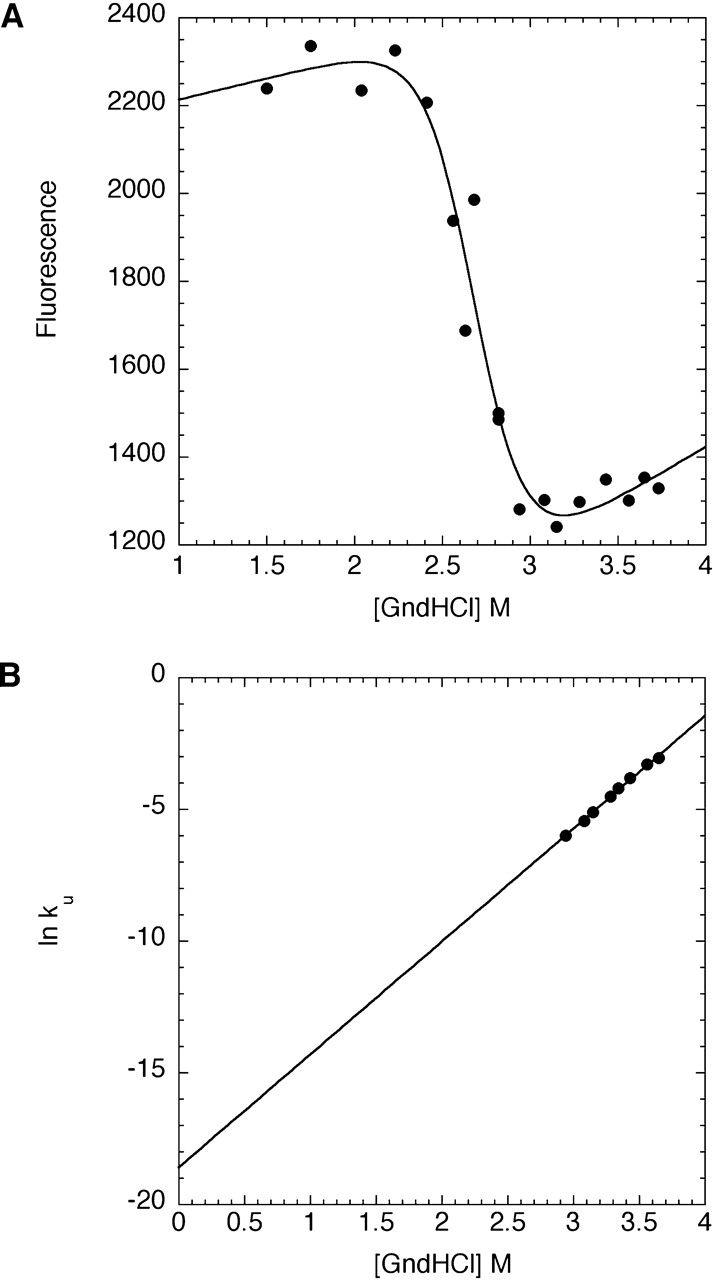
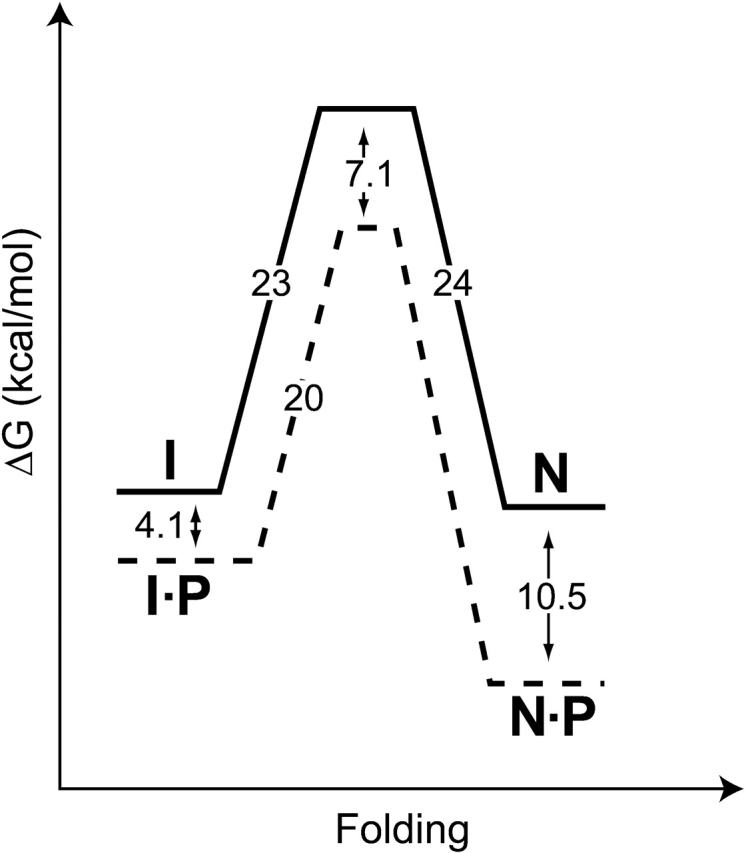
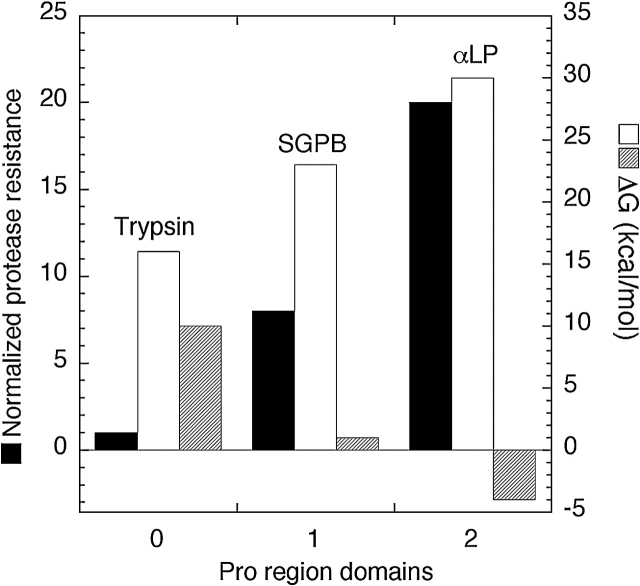
Like most extracellular bacterial proteases, Streptomyces griseus protease B (SGPB) and alpha-lytic protease (alphaLP) are synthesized with covalently attached pro regions necessary for their folding. In this article, we characterize the folding free energy landscape of SGPB and compare it to the folding landscapes of alphaLP and trypsin, a mammalian homolog that folds independently of its zymogen peptide. In contrast to the thermodynamically stable native state of trypsin, SGPB and alphaLP fold to native states that are thermodynamically marginally stable or unstable, respectively. Instead, their apparent stability arises kinetically, from unfolding free energy barriers that are both large and highly cooperative. The unique unfolding transitions of SGPB and alphaLP extend their functional lifetimes under highly degradatory conditions beyond that seen for trypsin; however, the penalty for evolving kinetic stability is remarkably large in that each factor of 2.4-8 in protease resistance is accompanied by a cost of ~10(5) in the spontaneous folding rate and ~5-9 kcal/mole in thermodynamic stability. These penalties have been overcome by the coevolution of increasingly effective pro regions to facilitate folding. Despite these costs, kinetic stability appears to be a potent mechanism for developing native-state properties that maximize protease longevity.
Figures

References
-
- Anderson, D.E., Peters, R.J., Wilk, B., and Agard, D. 1999. α-Lytic protease precursor: Characterization of a structured folding intermediate. Biochemistry 38 4728–4735. - PubMed
-
- Baker, D., Sohl, J.L., and Agard, D.A. 1992. A protein-folding reaction under kinetic control. Nature 356 263–265. - PubMed
-
- Bhattacharjya, S., Xu, P., and Ni, F. 2000. Sequence-specific 1H, 15N and 13C resonance assignments of the inhibitory prodomain of human furin. J. Biomol. NMR 16 275–276. - PubMed
-
- Bryan, P., Wang, L., Hoskins, J., Ruvinov, S., Strausberg, S., Alexander, P., Almog, O., Gilliland, G., and Gallagher, T. 1995. Catalysis of a protein folding reaction: Mechanistic implications of the 2.0 Å structure of the subtilisin–prodomain complex. Biochemistry 34 10310–10318. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
