

Local homology recognition and distance measures in linear time using compressed amino acid alphabets
- PMID: 14729922
- PMCID: PMC373290
- DOI: 10.1093/nar/gkh180
Local homology recognition and distance measures in linear time using compressed amino acid alphabets
Abstract
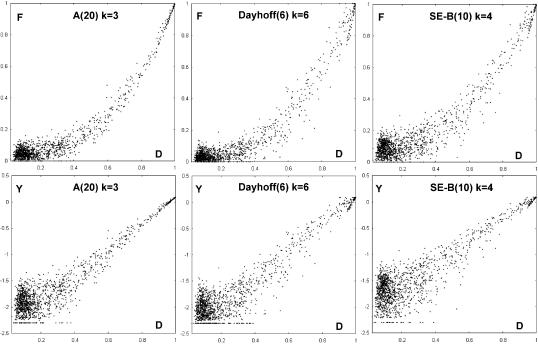
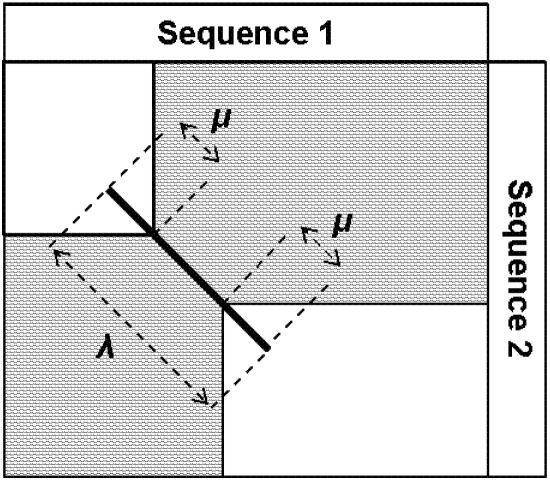
Methods for discovery of local similarities and estimation of evolutionary distance by identifying k-mers (contiguous subsequences of length k) common to two sequences are described. Given unaligned sequences of length L, these methods have O(L) time complexity. The ability of compressed amino acid alphabets to extend these techniques to distantly related proteins was investigated. The performance of these algorithms was evaluated for different alphabets and choices of k using a test set of 1848 pairs of structurally alignable sequences selected from the FSSP database. Distance measures derived from k-mer counting were found to correlate well with percentage identity derived from sequence alignments. Compressed alphabets were seen to improve performance in local similarity discovery, but no evidence was found of improvements when applied to distance estimates. The performance of our local similarity discovery method was compared with the fast Fourier transform (FFT) used in MAFFT, which has O(L log L) time complexity. The method for achieving comparable coverage to FFT is revealed here, and is more than an order of magnitude faster. We suggest using k-mer distance for fast, approximate phylogenetic tree construction, and show that a speed improvement of more than three orders of magnitude can be achieved relative to standard distance methods, which require alignments.
Figures

References
-
- Altschul S.F., Gish,W., Miller,W., Myers,E.E. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. - PubMed
-
- Sneath P.H.A. and Sokal,R.R. (1973) Numerical Taxonomy. Freeman, San Francisco, CA.
-
- Saitou N. and Nei,M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4, 406–425. - PubMed
-
- Durbin R., Eddy,S., Krogh,A. and Mitchison,G. (1998) Biological Sequence Analysis. Cambridge University Press, Cambridge, UK.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
