

Role for the pleckstrin homology domain-containing protein CKIP-1 in phosphatidylinositol 3-kinase-regulated muscle differentiation
- PMID: 14729969
- PMCID: PMC321442
- DOI: 10.1128/MCB.24.3.1245-1255.2004
Role for the pleckstrin homology domain-containing protein CKIP-1 in phosphatidylinositol 3-kinase-regulated muscle differentiation
Abstract
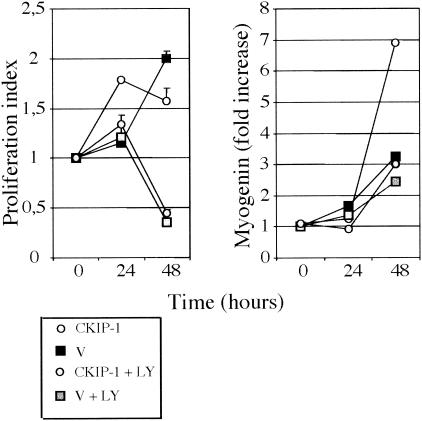
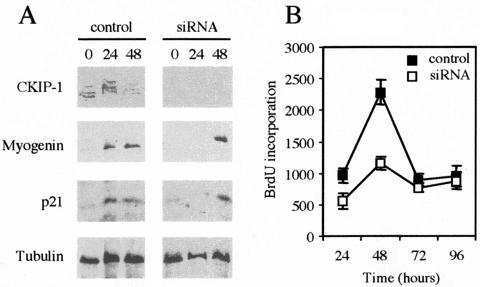
In this work, we report the implication of the pleckstrin homology (PH) domain-containing protein CKIP-1 in phosphatidylinositol 3-kinase (PI3-K)-regulated muscle differentiation. CKIP-1 is upregulated during muscle differentiation in C2C12 cells. We show that CKIP-1 binds to phosphatidylinositol 3-phosphate through its PH domain and localizes to the plasma membrane in a PI3-K-dependent manner. Activation of PI3-K by insulin or expression of an active form of PI3-K p110 induces a rapid translocation of CKIP-1 to the plasma membrane. Conversely, expression of the 3-phosphoinositide phosphatase myotubularin or PI3-K inhibition by LY294002, wortmannin, or mutant p85 abolishes CKIP-1 binding to the membrane. Upon induction of differentiation in low-serum medium, CKIP-1 overexpression in C2C12 myoblasts first promotes proliferation and then stimulates the expression of myogenin and cell fusion in a manner reminiscent of the dual positive effect of insulin-like growth factors on muscle cells. Interference with the PI3-K pathway impedes the effect of CKIP-1 on C2C12 cell differentiation. Finally, silencing of CKIP-1 by RNA interference abolishes proliferation and delays myogenin expression. Altogether, these data strongly implicate CKIP-1 as a new component of PI3-K signaling in muscle differentiation.
Figures







References
-
- Alessi, D. R., S. R. James, C. P. Downes, A. B. Holmes, P. R. Gaffney, C. B. Reese, and P. Cohen. 1997. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B alpha. Curr. Biol. 7:261-269. - PubMed
-
- Andreucci, J. J., D. Grant, D. M. Cox, L. K. Tomc, R. Prywes, D. J. Goldhamer, N. Rodrigues, P. A. Bedard, and J. C. McDermott. 2002. Composition and function of AP-1 transcription complexes during muscle cell differentiation. J. Biol. Chem. 277:16426-16432. - PubMed
-
- Bosc, D. G., K. C. Graham, R. B. Saulnier, C. Zhang, D. Prober, R. D. Gietz, and D. W. Litchfield. 2000. Identification and characterization of CKIP-1, a novel pleckstrin homology domain-containing protein that interacts with protein kinase CK2. J. Biol. Chem. 275:14295-142306. - PubMed
-
- Calera, M. R., and P. F. Pilch. 1998. Induction of Akt-2 correlates with differentiation in Sol8 muscle cells. Biochem. Biophys. Res. Commun. 251:835-841. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases