

Similarities and differences in genome-wide expression data of six organisms
- PMID: 14737187
- PMCID: PMC300882
- DOI: 10.1371/journal.pbio.0020009
Similarities and differences in genome-wide expression data of six organisms
Abstract
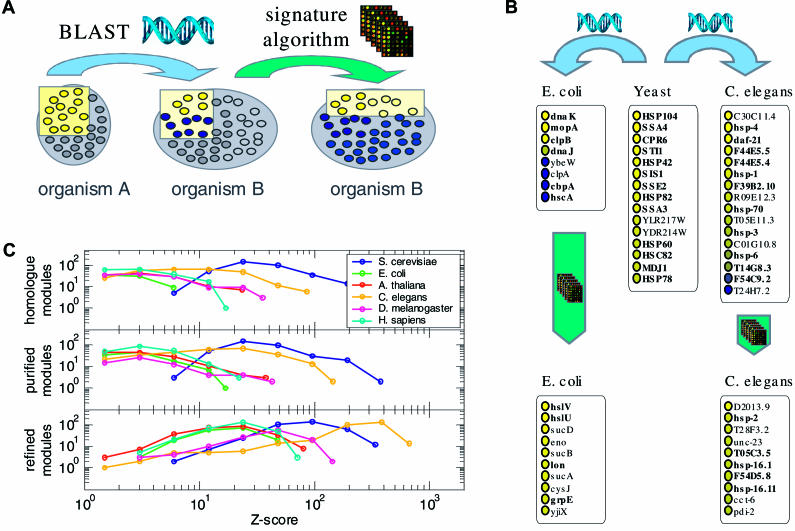
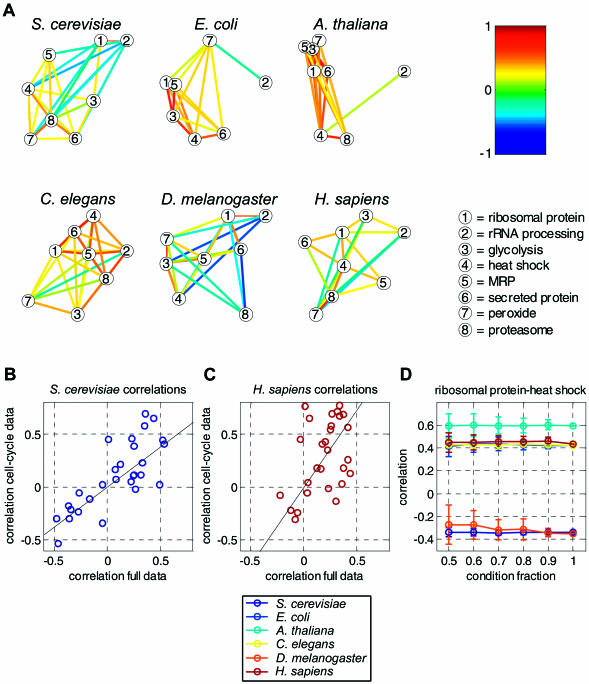
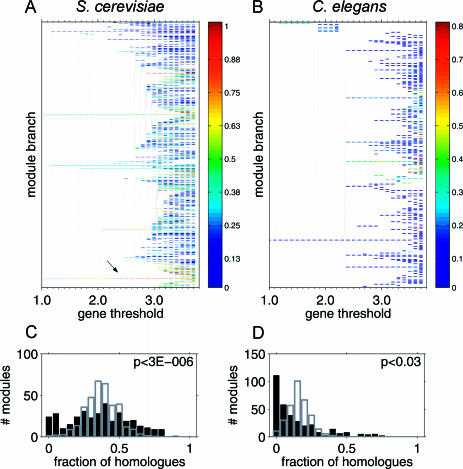
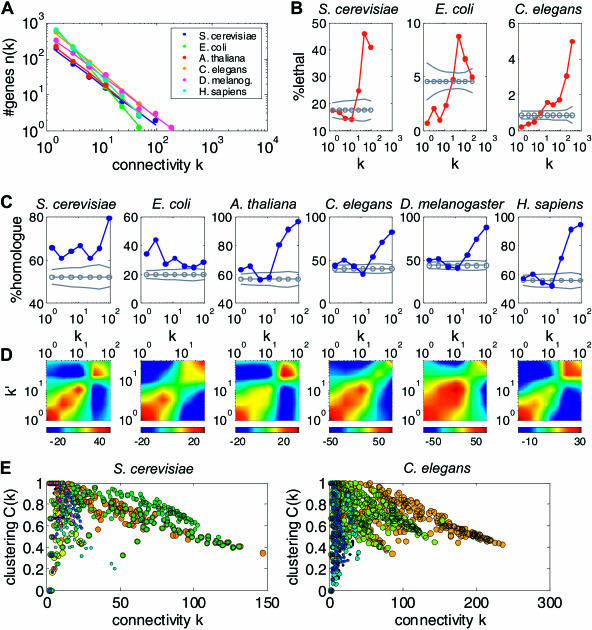
Comparing genomic properties of different organisms is of fundamental importance in the study of biological and evolutionary principles. Although differences among organisms are often attributed to differential gene expression, genome-wide comparative analysis thus far has been based primarily on genomic sequence information. We present a comparative study of large datasets of expression profiles from six evolutionarily distant organisms: S. cerevisiae, C. elegans, E. coli, A. thaliana, D. melanogaster, and H. sapiens. We use genomic sequence information to connect these data and compare global and modular properties of the transcription programs. Linking genes whose expression profiles are similar, we find that for all organisms the connectivity distribution follows a power-law, highly connected genes tend to be essential and conserved, and the expression program is highly modular. We reveal the modular structure by decomposing each set of expression data into coexpressed modules. Functionally related sets of genes are frequently coexpressed in multiple organisms. Yet their relative importance to the transcription program and their regulatory relationships vary among organisms. Our results demonstrate the potential of combining sequence and expression data for improving functional gene annotation and expanding our understanding of how gene expression and diversity evolved.
Conflict of interest statement
The authors have declared that no conflicts of interest exist.
Figures
References
-
- Albert R, Barabasi A-L. Statistical mechanics of complex networks. Rev Mod Phys v. 2002;74:47–97.
-
- Arbeitman MN, Furlong EE, Imam F, Johnson E, Null BH, et al. Gene expression during the life cycle of Drosophila melanogaster . Science. 2002;297:2270–2275. - PubMed
-
- Barbasi A-L, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
