

Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously
- PMID: 14739232
- PMCID: PMC373371
- DOI: 10.1093/nar/gnh014
Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously
Abstract
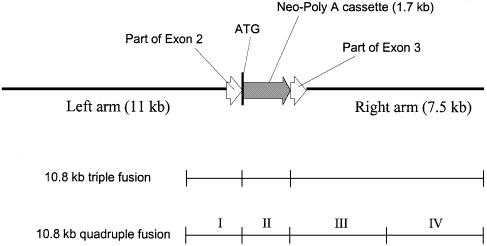
A procedure for precise assembly of linear DNA constructs as long as 20 kb is proposed. The method, which we call long multiple fusion, has been used to assemble up to four fragments simultaneously (for a 10.8 kb final product), offering an additional improvement on the combination of long PCR and overlap extension PCR. The method is based on Pfu polymerase mix, which has a proofreading activity. We successfully assembled (and confirmed by sequencing) seven different linear constructs ranging from 3 to 20 kb, including two 20 kb products (from fragments of 11, 1.7 and 7.5 kb), two 10.8 kb constructs, and two constructs of 6.1 and 6.2 kb, respectively. Accuracy of the PCR fusion is greater than or equal to one error per 6.6 kb, which is consistent with the expected error rate of the PCR mix. The method is expected to facilitate various kinds of complex genetic engineering projects that require precise in-frame assembly of multiple fragments, such as somatic cell knockout in human cells or creation of whole genomes of viruses for vaccine research.
Figures











References
-
- Pont-Kingdon G. (1997) Creation of chimeric junctions, deletions, and insertions by PCR. Methods Mol. Biol., 67, 167–172. - PubMed
-
- Sedivy J.M., Vogelstein,B., Liber,H.L., Hendrickson,E.A. and Rosmarin,A. (1999) Gene targeting in human cells without isogenic DNA. Science, 283, 9a.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
