

Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II
- PMID: 14740321
- PMCID: PMC1181933
- DOI: 10.1086/381685
Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II
Erratum in
- Am J Hum Genet. 2004 May;74(5):1080
Abstract
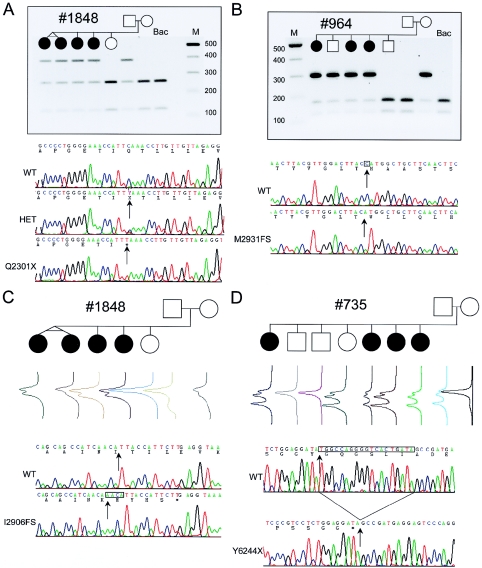
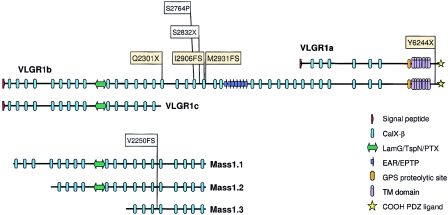
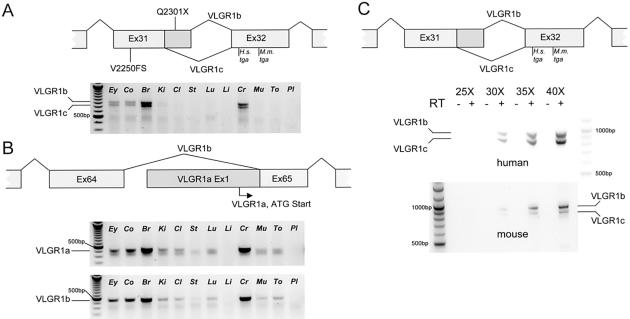
Usher syndrome type II (USH2) is a genetically heterogeneous autosomal recessive disorder with at least three genetic subtypes (USH2A, USH2B, and USH2C) and is classified phenotypically as congenital hearing loss and progressive retinitis pigmentosa. The VLGR1 (MASS1) gene in the 5q14.3-q21.1 USH2C locus was considered a likely candidate on the basis of its protein motif structure and expressed-sequence-tag representation from both cochlear and retinal subtracted libraries. Denaturing high-performance liquid chromatography and direct sequencing of polymerase-chain-reaction products amplified from 10 genetically independent patients with USH2C and 156 other patients with USH2 identified four isoform-specific VLGR1 mutations (Q2301X, I2906FS, M2931FS, and T6244X) from three families with USH2C, as well as two sporadic cases. All patients with VLGR1 mutations are female, a significant deviation from random expectations. The ligand(s) for the VLGR1 protein is unknown, but on the basis of its potential extracellular and intracellular protein-protein interaction domains and its wide mRNA expression profile, it is probable that VLGR1 serves diverse cellular and signaling processes. VLGR1 mutations have been previously identified in both humans and mice and are associated with a reflex-seizure phenotype in both species. The identification of additional VLGR1 mutations to test whether a phenotype/genotype correlation exists, akin to that shown for other Usher syndrome disease genes, is warranted.
Figures
References
Electronic-Database Information
-
- National Eye Institute Gene Bank, http://neibank.nei.nih.gov/libs/NbLib0013/NbLib0013.shtml (for human retina) and http://neibank.nei.nih.gov/libs/NbLib0011/NbLib0011.shtml (for human fetal cochlea)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for USH2, USH1D, USH1F, USH2C, and FEB4)
-
- UCSC Genome Browser, http://genome.cse.ucsc.edu/ (for human VLGR1, AF435925; mouse Vlgr1, AF435926; and VLGR1 genomic DNA, NT_028179)
References
-
- Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CR, Lowry RB, Knaus R, Van Laer L, Bernier FP, Schwartz S, Lee C, Morton CC, Mullins RF, Ramesh A, Van Camp G, Hageman GS, Woychik RP, Smith RJ (2001) Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet 10:1709–171810.1093/hmg/10.16.1709 - DOI - PubMed
-
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, Barnes PD, O’Brien RE, Farndon PA, Sowden J, Liu XZ, Scanlan MJ, Malcolm S, Dunne MJ, Aynsley-Green A, Glaser B (2000) A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat Genet 26:56–6010.1038/79178 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
