

Functional analysis of the cag pathogenicity island in Helicobacter pylori isolates from patients with gastritis, peptic ulcer, and gastric cancer
- PMID: 14742552
- PMCID: PMC321631
- DOI: 10.1128/IAI.72.2.1043-1056.2004
Functional analysis of the cag pathogenicity island in Helicobacter pylori isolates from patients with gastritis, peptic ulcer, and gastric cancer
Abstract
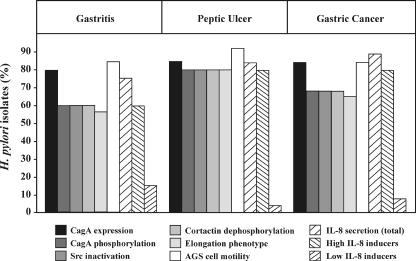
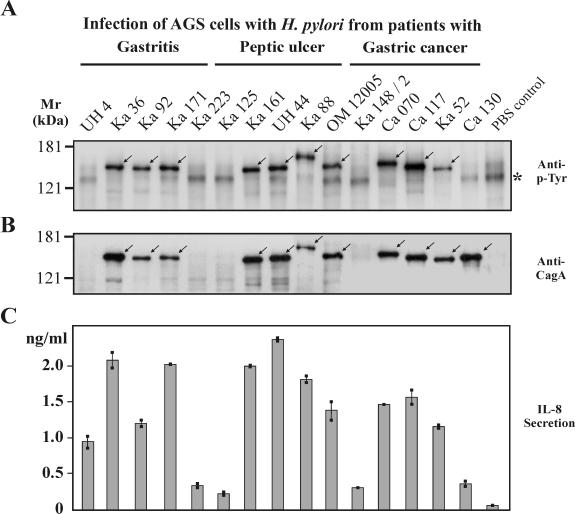
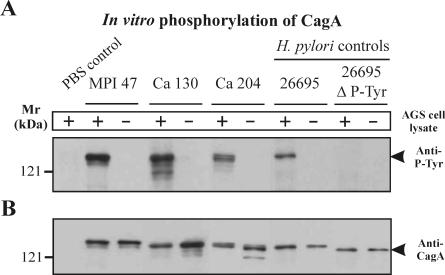
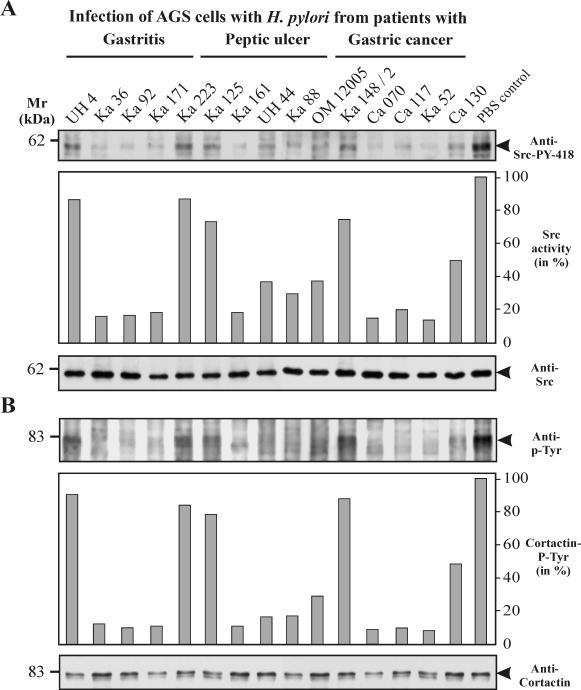
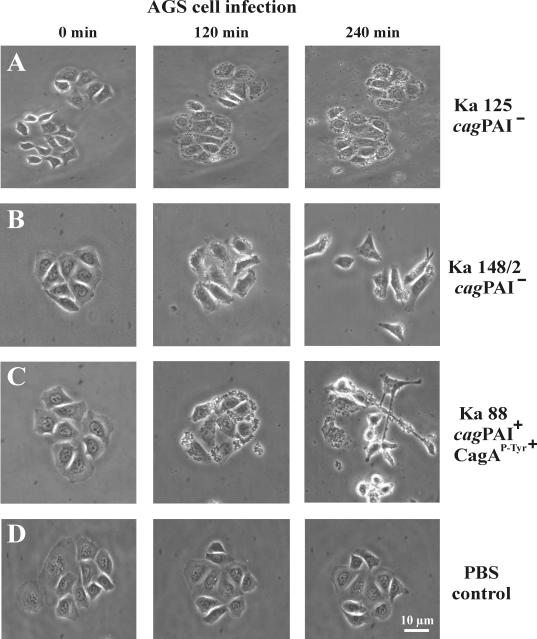
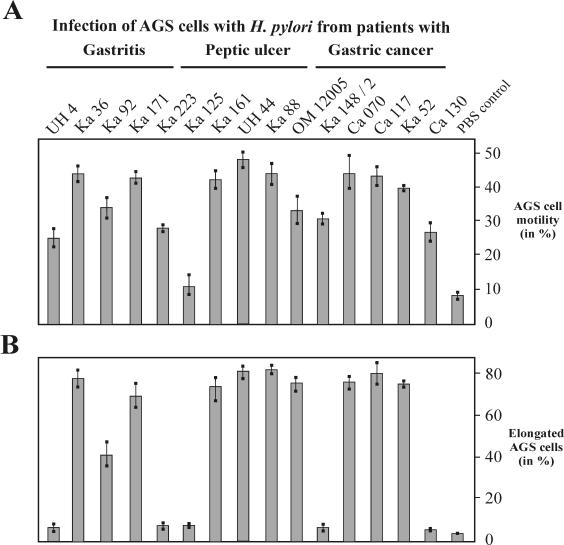
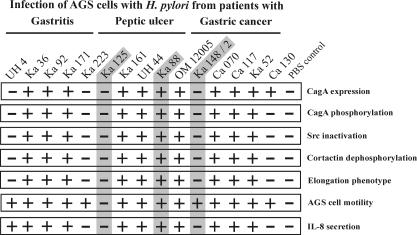
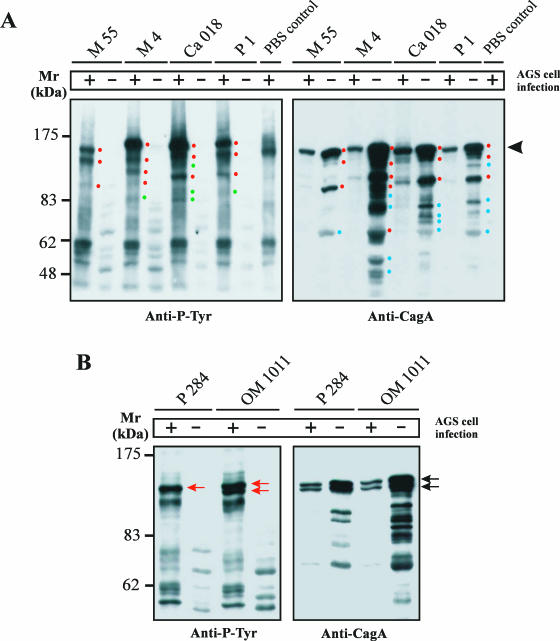
Helicobacter pylori is the causative agent of a variety of gastric diseases, but the clinical relevance of bacterial virulence factors is still controversial. Virulent strains carrying the cag pathogenicity island (cagPAI) are thought to be key players in disease development. Here, we have compared cagPAI-dependent in vitro responses in H. pylori isolates obtained from 75 patients with gastritis, peptic ulcer, and gastric cancer (n = 25 in each group). AGS gastric epithelial cells were infected with each strain and assayed for (i) CagA expression, (ii) translocation and tyrosine phosphorylation of CagA, (iii) c-Src inactivation, (iv) cortactin dephosphorylation, (v) induction of actin cytoskeletal rearrangements associated with cell elongation, (vi) induction of cellular motility, and (vii) secretion of interleukin-8. Interestingly, we found high but similar prevalences of all of these cagPAI-dependent host cell responses (ranging from 56 to 80%) among the various groups of patients. This study revealed CagA proteins with unique features, CagA subspecies of various sizes, and new functional properties for the phenotypic outcomes. We further showed that induction of AGS cell motility and elongation are two independent processes. Our data corroborate epidemiological studies, which indicate a significant association of cagPAI presence and functionality with histopathological findings in gastritis, peptic ulcer, and gastric cancer patients, thus emphasizing the importance of the cagPAI for the pathogenicity of H. pylori. Nevertheless, we found no significant association of the specific H. pylori-induced responses with any particular patient group. This may indicate that the determination of disease development is highly complex and involves multiple bacterial and/or host factors.
Figures
References
-
- Akanuma, M., S. Maeda, K. Ogura, Y. Mitsuno, Y. Hirata, T. Ikenoue, M. Otsuka, T. Watanabe, Y. Yamaji, H. Yoshida, T. Kawabe, Y. Shiratori, and M. Omata. 2002. The evaluation of putative virulence factors of Helicobacter pylori for gastroduodenal disease by use of a short-term Mongolian gerbil infection model. J. Infect. Dis. 185:341-347. - PubMed
-
- Akopyants, N. S., S. W. Clifton, D. Kersulyte, J. E. Crabtree, B. E. Youree, C. A. Reece, N. O. Bukanov, E. S. Drazek, B. A. Roe, and D. E. Berg. 1998. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol. Microbiol. 28:37-53. - PubMed
-
- Ando, T., K. Kusugami, M. Ohsuga, M. Shinoda, M. Sakakibara, H. Saito, A. Fukatsu, S. Ichiyama, and M. Ohta. 1996. Interleukin-8 activity correlates with histological severity in Helicobacter pylori-associated antral gastritis. Am. J. Gastroenterol. 91:1150-1156. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
