

Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta
- PMID: 14744935
- PMCID: PMC338280
- DOI: 10.1101/gad.1165804
Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta
Abstract
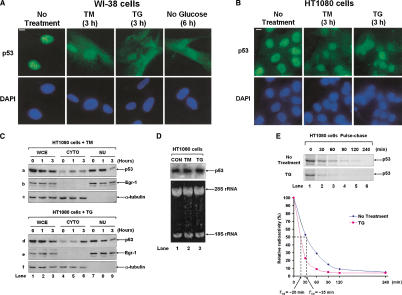
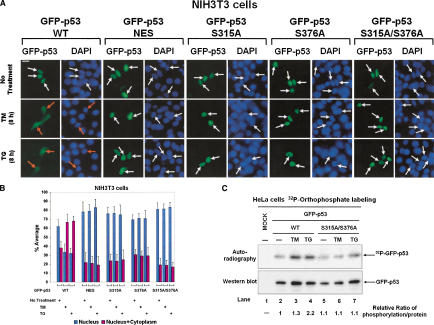
The tumor suppressor p53, a sensor of multiple forms of cellular stress, is regulated by post-translational mechanisms to induce cell-cycle arrest, senescence, or apoptosis. We demonstrate that endoplasmic reticulum (ER) stress inhibits p53-mediated apoptosis. The mechanism of inhibition involves the increased cytoplasmic localization of p53 due to phosphorylation at serine 315 and serine 376, which is mediated by glycogen synthase kinase-3 beta (GSK-3beta). ER stress induces GSK-3beta binding to p53 in the nucleus and enhances the cytoplasmic localization of the tumor suppressor. Inhibition of apoptosis caused by ER stress requires GSK-3beta and does not occur in cells expressing p53 with mutation(s) of serine 315 and/or serine 376 to alanine(s). As a result of the increased cytoplasmic localization, ER stress prevents p53 stabilization and p53-mediated apoptosis upon DNA damage. It is concluded that inactivation of p53 is a protective mechanism utilized by cells to adapt to ER stress.
Figures




Comment in
-
p53 and stress in the ER.Genes Dev. 2004 Feb 1;18(3):241-4. doi: 10.1101/gad.1181704. Genes Dev. 2004. PMID: 14871924 No abstract available.
References
-
- Aloni-Grinstein R., Zan-Bar, I., Alboum, I., Goldfinger, N., and Rotter, V. 1993. Wild type p53 functions as a control protein in the differentiation pathway of the B-cell lineage. Oncogene 8: 3297-3305. - PubMed
-
- Appella E. and Anderson, C.W. 2001. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 268: 2764-2772. - PubMed
-
- Aridor M. and Balch, W.E. 1999. Integration of endoplasmic reticulum signaling in health and disease. Nat. Med. 5: 745-751. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous