

Analysis of functional motions in Brownian molecular machines with an efficient block normal mode approach: myosin-II and Ca2+ -ATPase
- PMID: 14747312
- PMCID: PMC1303924
- DOI: 10.1016/S0006-3495(04)74152-1
Analysis of functional motions in Brownian molecular machines with an efficient block normal mode approach: myosin-II and Ca2+ -ATPase
Abstract
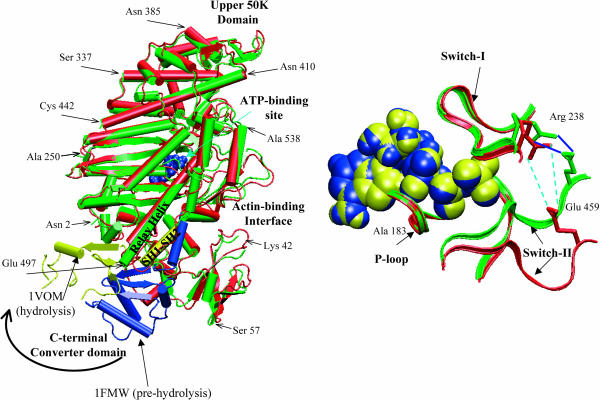
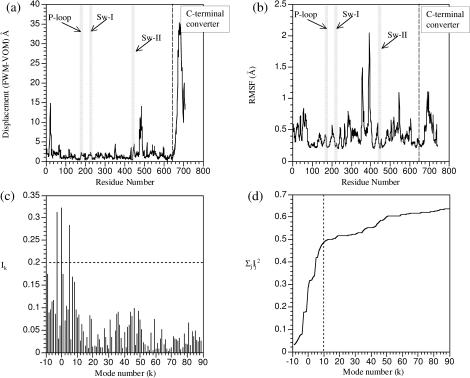
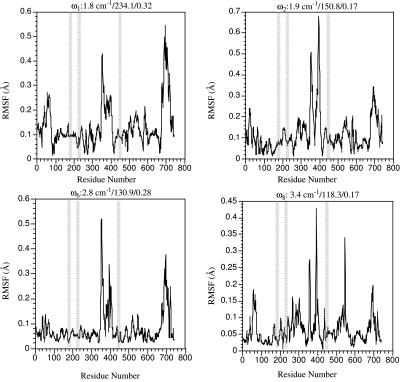
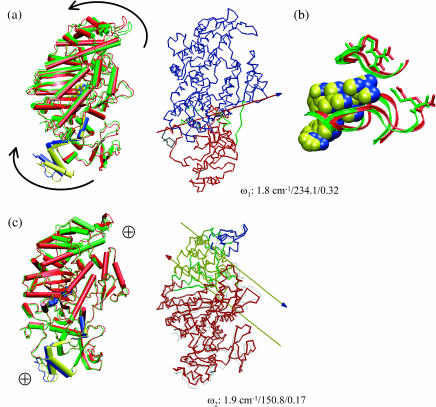
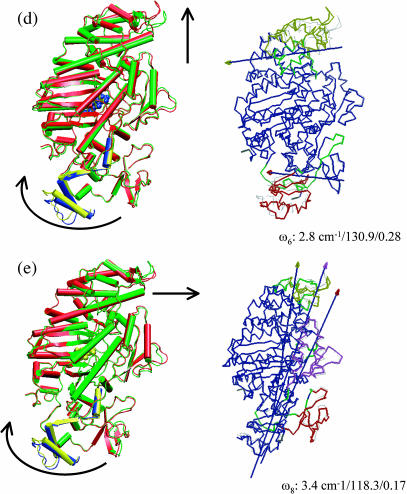
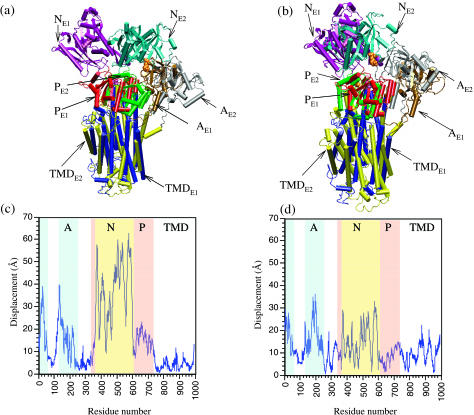
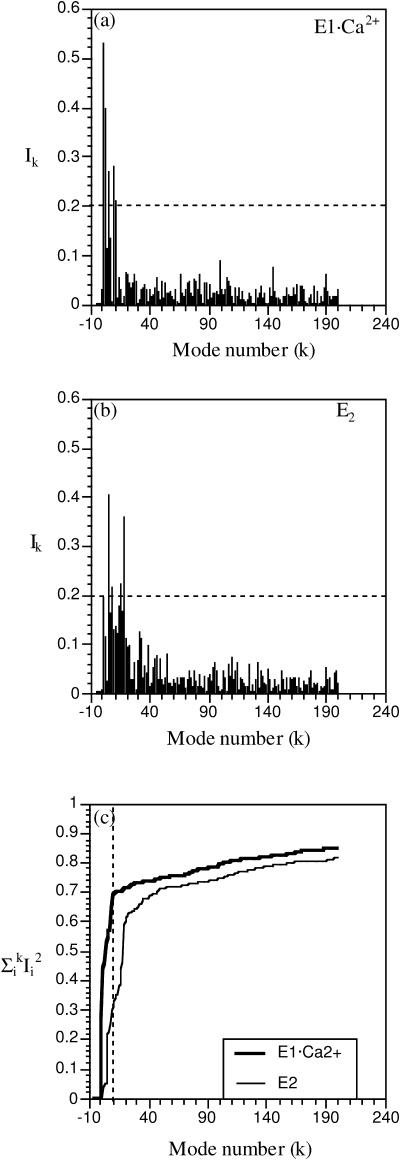
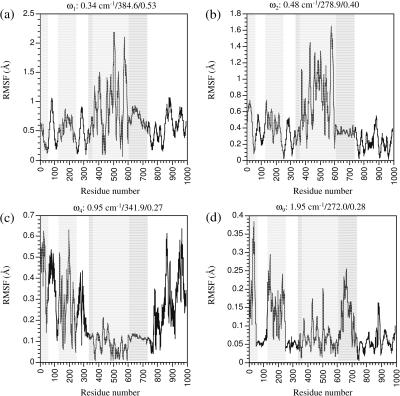
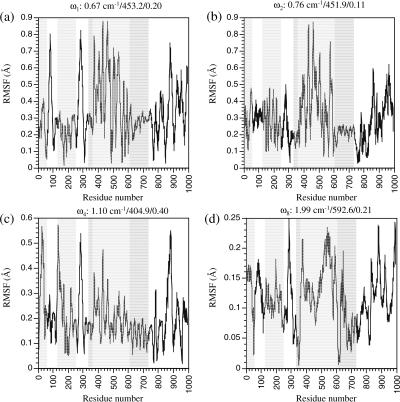
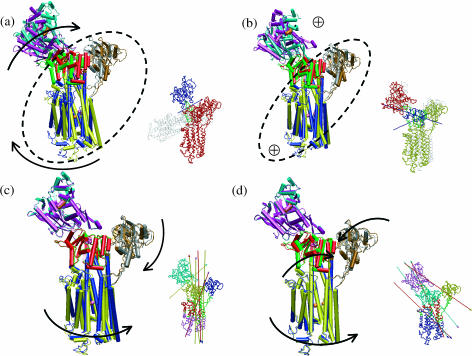
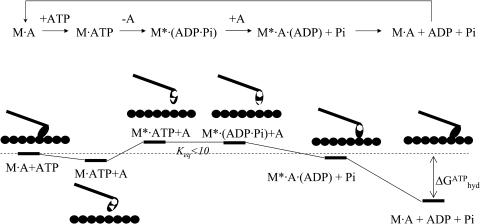
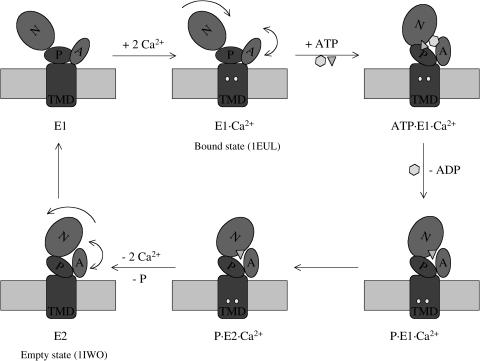
The structural flexibilities of two molecular machines, myosin and Ca(2+)-ATPase, have been analyzed with normal mode analysis and discussed in the context of their energy conversion functions. The normal mode analysis with physical intermolecular interactions was made possible by an improved implementation of the block normal mode (BNM) approach. The BNM results clearly illustrated that the large-scale conformational transitions implicated in the functional cycles of the two motor systems can be largely captured with a small number of low-frequency normal modes. Therefore, the results support the idea that structural flexibility is an essential part of the construction principle of molecular motors through evolution. Such a feature is expected to be more prevalent in motor proteins than in simpler systems (e.g., signal transduction proteins) because in the former, large-scale conformational transitions often have to occur before the chemical events (e.g., ATP hydrolysis in myosin and ATP binding/phosphorylation in Ca(2+)-ATPase). This highlights the importance of Brownian motions associated with the protein domains that are involved in the functional transitions; in this sense, Brownian molecular machines is an appropriate description of molecular motors, although the normal mode results do not address the origin of the ratchet effect. The results also suggest that it might be more appropriate to describe functional transitions in some molecular motors as intrinsic elastic motions modulating local structural changes in the active site, which in turn gets stabilized by the subsequent chemical events, in contrast with the conventional idea of local changes somehow getting amplified into larger-scale motions. In the case of myosin, for example, we favor the idea that Brownian motions associated with the flexible converter propagates to the Switch I/II region, where the salt-bridge formation gets stabilized by ATP hydrolysis, in contrast with the textbook notion that ATP hydrolysis drives the converter motion. Another useful aspect of the BNM results is that selected low-frequency normal modes have been identified to form a set of collective coordinates that can be used to characterize the progress of a significant fraction of large-scale conformational transitions. Therefore, the present normal mode analysis has provided a stepping-stone toward more elaborate microscopic simulations for addressing critical issues in free energy conversions in molecular machines, such as the coupling and the causal relationship between collective motions and essential local changes at the catalytic active site where ATP hydrolysis occurs.
Figures


Similar articles
-
A coarse-grained normal mode approach for macromolecules: an efficient implementation and application to Ca(2+)-ATPase.Biophys J. 2002 Nov;83(5):2457-74. doi: 10.1016/S0006-3495(02)75257-0. Biophys J. 2002. PMID: 12414680 Free PMC article.
-
Brownian ratchet models of molecular motors.Cell Biochem Biophys. 2003;38(2):191-214. doi: 10.1385/CBB:38:2:191. Cell Biochem Biophys. 2003. PMID: 12777714 Review.
-
The principal motions involved in the coupling mechanism of the recovery stroke of the myosin motor.J Mol Biol. 2007 Mar 23;367(2):591-602. doi: 10.1016/j.jmb.2006.12.058. Epub 2006 Dec 23. J Mol Biol. 2007. PMID: 17275022
-
Simulations of the myosin II motor reveal a nucleotide-state sensing element that controls the recovery stroke.J Mol Biol. 2006 Aug 18;361(3):604-16. doi: 10.1016/j.jmb.2006.06.022. Epub 2006 Jun 30. J Mol Biol. 2006. PMID: 16859703
-
Folding funnels and conformational transitions via hinge-bending motions.Cell Biochem Biophys. 1999;31(2):141-64. doi: 10.1007/BF02738169. Cell Biochem Biophys. 1999. PMID: 10593256 Review.
Cited by
-
The structural dynamics of macromolecular processes.Curr Opin Cell Biol. 2009 Feb;21(1):97-108. doi: 10.1016/j.ceb.2009.01.022. Epub 2009 Feb 14. Curr Opin Cell Biol. 2009. PMID: 19223165 Free PMC article. Review.
-
Allosteric activation transitions in enzymes and biomolecular motors: insights from atomistic and coarse-grained simulations.Top Curr Chem. 2013;337:139-64. doi: 10.1007/128_2012_409. Top Curr Chem. 2013. PMID: 23468286 Free PMC article. Review.
-
Probing the local dynamics of nucleotide-binding pocket coupled to the global dynamics: myosin versus kinesin.Biophys J. 2005 Jul;89(1):167-78. doi: 10.1529/biophysj.105.063305. Epub 2005 May 6. Biophys J. 2005. PMID: 15879477 Free PMC article.
-
Mechanochemical coupling in the myosin motor domain. II. Analysis of critical residues.PLoS Comput Biol. 2007 Feb 16;3(2):e23. doi: 10.1371/journal.pcbi.0030023. Epub 2006 Dec 22. PLoS Comput Biol. 2007. PMID: 17305418 Free PMC article.
-
Can conformational change be described by only a few normal modes?Biophys J. 2006 Mar 1;90(5):1583-93. doi: 10.1529/biophysj.105.070045. Epub 2005 Dec 16. Biophys J. 2006. PMID: 16361336 Free PMC article.
References
-
- Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. D. Watson. 1994. Molecular Biology of the Cell, 3rd Ed. Garland Publishing, New York and London, UK.
-
- Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Prot. Struct. Funct. Gen. 17:412–425. - PubMed
-
- Andersen, J. P. 1995. Dissection of the functional domains of the sarcoplasmic reticulum Ca2+-ATPase by site-directed mutagenesis. Biosci. Rep. 15:243–261. - PubMed
-
- Andersen, J. P., J. D. Clausen, A. P. Einholm, and B. Vilsen. 2003. Mutagenesis of residues involved in control of the Ca2+ entry pathway and conformational changes associated with Ca2+ binding in the SR Ca2+-ATPase. Ann. NY Acad. Sci. 986:72–81. - PubMed
-
- Andersen, J. P., and T. Sorensen. 1996. Site-directed mutagenesis studies of energy coupling in the sarcoplasmic reticulum Ca2+-ATPase. Biochim. Biophys. Acta Bioenerg. 1275:118–122. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
