

Alpha0 thalassaemia as a result of a novel 11.1 kb deletion eliminating both of the duplicated alpha globin genes
- PMID: 14747442
- PMCID: PMC1770193
- DOI: 10.1136/jcp.2003.12856
Alpha0 thalassaemia as a result of a novel 11.1 kb deletion eliminating both of the duplicated alpha globin genes
Abstract
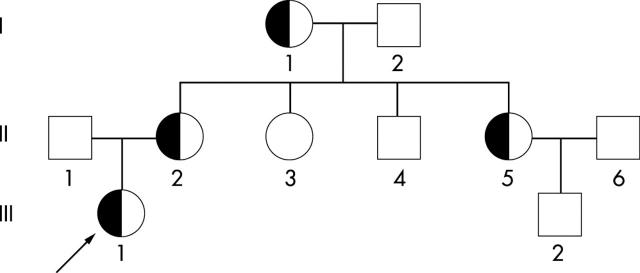
Aims: To characterise a novel 11.1 kb deletion that eliminated both of the duplicated alpha globin genes, giving rise to a typical alpha0 thalassaemia phenotype in four carriers from a Chinese family.
Methods: Haematological investigations were carried out on all family members. The seven common forms of alpha thalassaemia were screened for by the polymerase chain reaction (PCR) and Southern blotting was used to analyse the alpha globin gene cluster. DNA sequence analysis of the entire alpha1 and alpha1 globin gene region was carried out and reverse transcription (RT)-PCR was used to investigate the transcription levels of the alpha and beta globin genes.
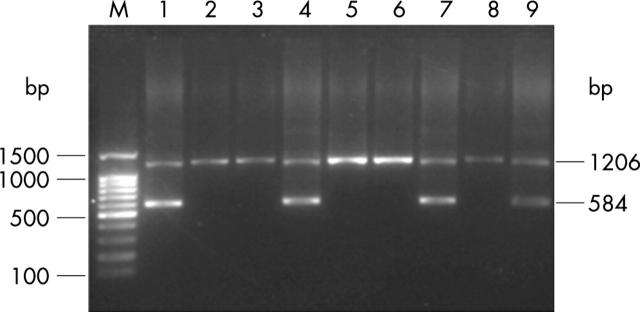
Results: The breakpoints were found to lie between coordinates 31695-31724 and 42846-42867 of the alpha globin gene cluster (NG_000006), with a total of about 11,135 nucleotides deleted. These sequences are involved in (CA)n repeats, suggesting a homologous recombination event. RT-PCR analysis gave a transcription level of the alpha globin gene in heterozygotes comparable with that of SEA deletion heterozygotes, confirming no output of alpha globin from the linked pair of alpha globin genes. The heterozygosity for this novel deletion was confirmed by PCR diagnosis in all four carriers from this family.
Conclusions: This rare mutation constitutes an additional heterogeneous defect causing alpha thalassaemia in the Chinese population.
Figures

References
-
- Higgs DR, Thein SL, Wood WG. The biology of the thalassemias. In: Weatherall DJ, Clegg JB, eds. The thalassemia syndromes, 4th ed. Massachusetts: Blackwell Science, 2001:65–237.
-
- Cao A, Rosatelli MC, Monni G, et al. Screening for thalassemia: a model of success. Obstet Gynecol Clin North Am 2002;29:305vi–28vii. - PubMed
-
- Ko TM, Xu X. Molecular study and prenatal diagnosis of alpha- and beta-thalassemias in Chinese. J Formos Med Assoc 1998;97:5–15. - PubMed
-
- Zeng YT, Huang SZ. Alpha-globin gene organisation and prenatal diagnosis of alpha-thalassaemia in Chinese. Lancet 1985;1:304–7. - PubMed
-
- Liang R, Liang S, Jiang NH, et al. Alpha and beta thalassaemia among Chinese children in Guangxi Province, P.R. China: molecular and haematological characterization. Br J Haematol 1994;86:351–4. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources