

Innate cellular response to virus particle entry requires IRF3 but not virus replication
- PMID: 14747536
- PMCID: PMC369475
- DOI: 10.1128/jvi.78.4.1706-1717.2004
Innate cellular response to virus particle entry requires IRF3 but not virus replication
Abstract
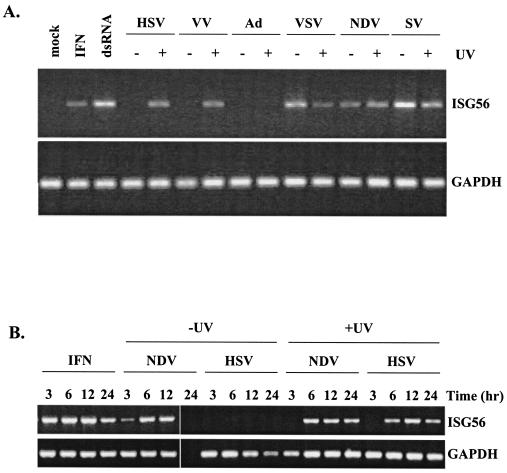
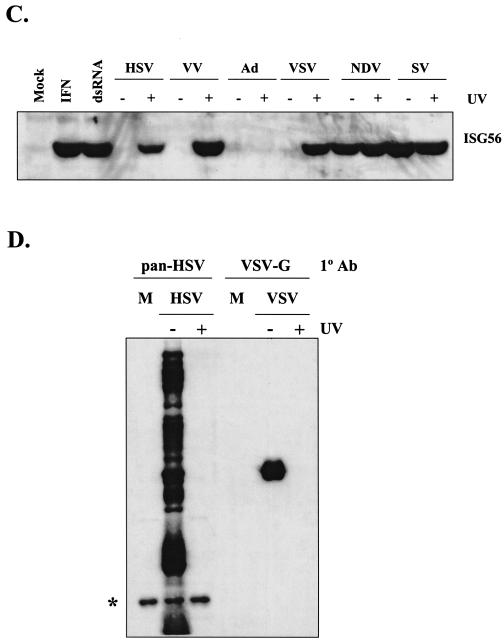
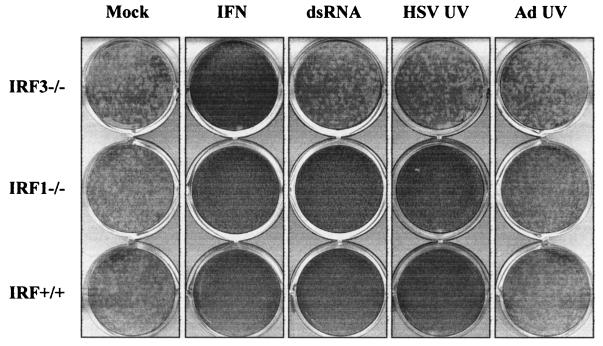
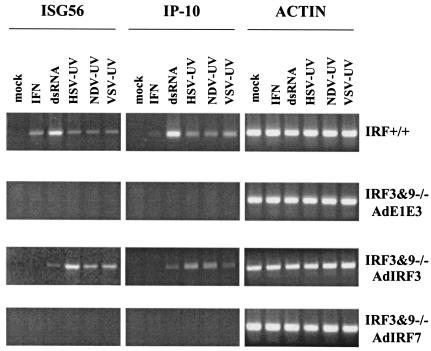
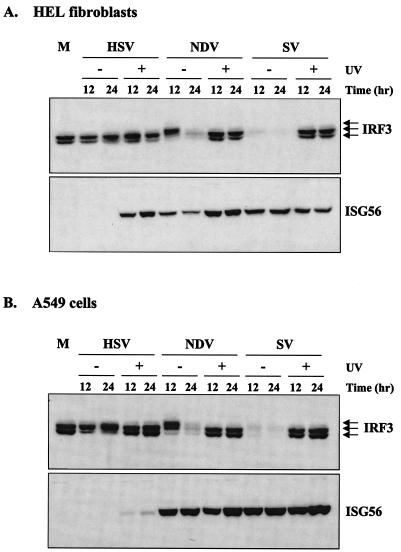
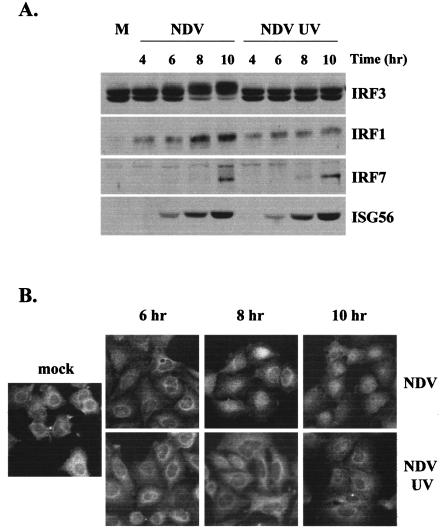
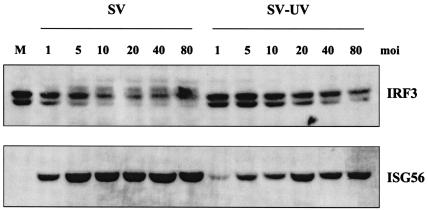
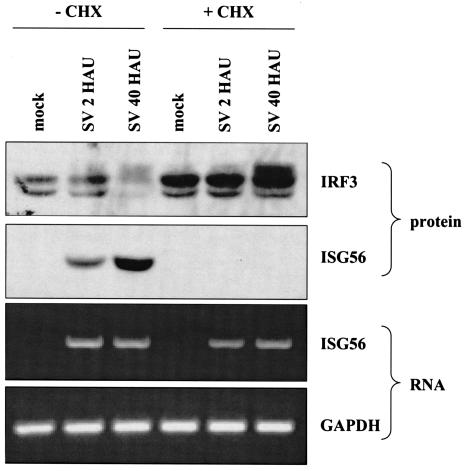
Mammalian cells respond to virus infections by eliciting both innate and adaptive immune responses. One of the most effective innate antiviral responses is the production of alpha/beta interferon and the subsequent induction of interferon-stimulated genes (ISGs), whose products collectively limit virus replication and spread. Following viral infection, interferon is produced in a biphasic fashion that involves a number of transcription factors, including the interferon regulatory factors (IRFs) 1, 3, 7, and 9. In addition, virus infection has been shown to directly induce ISGs in the absence of prior interferon production through the activation of IRF3. This process is believed to require virus replication and results in IRF3 hyperphosphorylation, nuclear localization, and proteasome-mediated degradation. Previously, we and others demonstrated that herpes simplex virus type 1 (HSV-1) induces ISGs and an antiviral response in fibroblasts in the absence of both interferon production and virus replication. In this report, we show that the entry of enveloped virus particles from diverse virus families elicits a similar innate response. This process requires IRF3, but not IRF1, IRF7, or IRF9. Following virus replication, the large DNA viruses HSV-1 and vaccinia virus effectively inhibit ISG mRNA accumulation, whereas the small RNA viruses Newcastle disease virus, Sendai virus, and vesicular stomatitis virus do not. In addition, we found that IRF3 hyperphosphorylation and degradation do not correlate with ISG and antiviral state induction but instead serve as a hallmark of productive virus replication, particularly following a high-multiplicity infection. Collectively, these data suggest that virus entry triggers an innate antiviral response mediated by IRF3 and that subsequent virus replication results in posttranslational modification of IRF3, such as hyperphosphorylation, depending on the nature of the incoming virus.
Figures
References
-
- Bandyopadhyay, S. K., G. T. Leonaard, T. Bandyopadhyay, G. R. Stark, and G. C. Sen. 1995. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem. 270:19624-19629. - PubMed
-
- Barnes, B., B. Lubyova, and P. M. Pitha. 2002. On the role of IRF in host defense. J. Interferon Cytokine Res. 22:59-71. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
