

Mutant PrPSc conformers induced by a synthetic peptide and several prion strains
- PMID: 14747574
- PMCID: PMC369494
- DOI: 10.1128/jvi.78.4.2088-2099.2004
Mutant PrPSc conformers induced by a synthetic peptide and several prion strains
Abstract
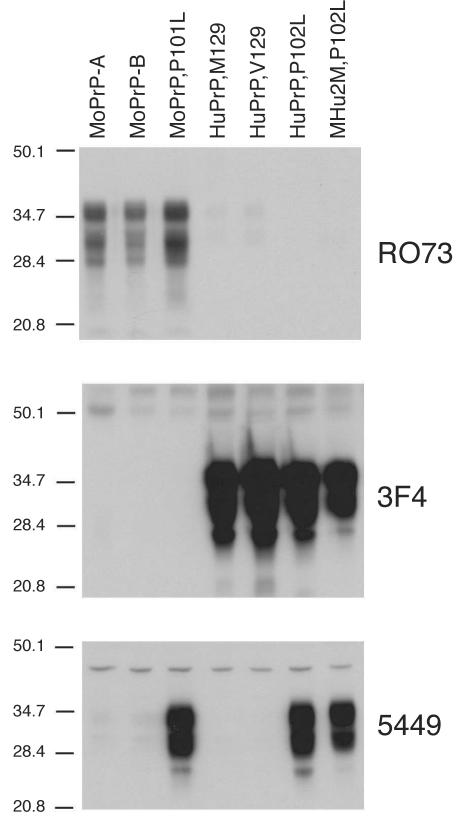
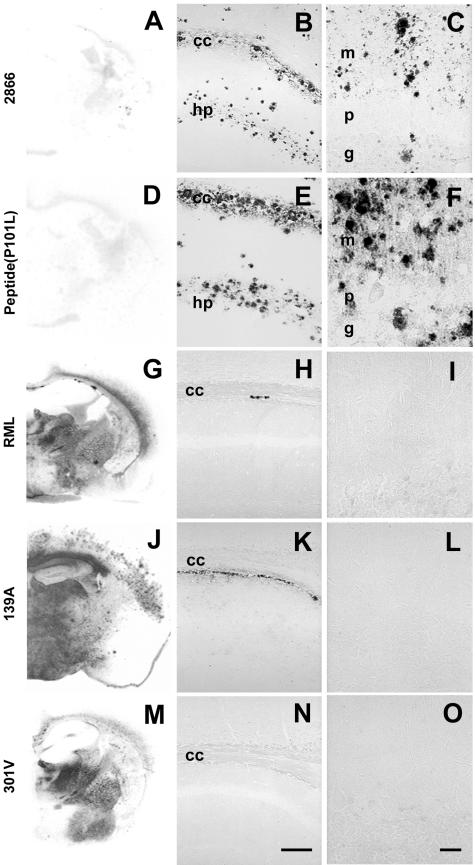
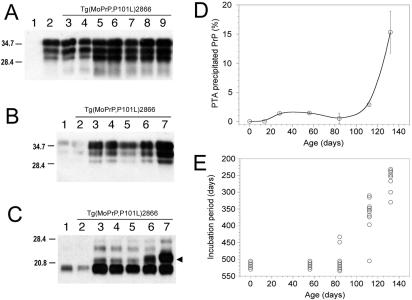
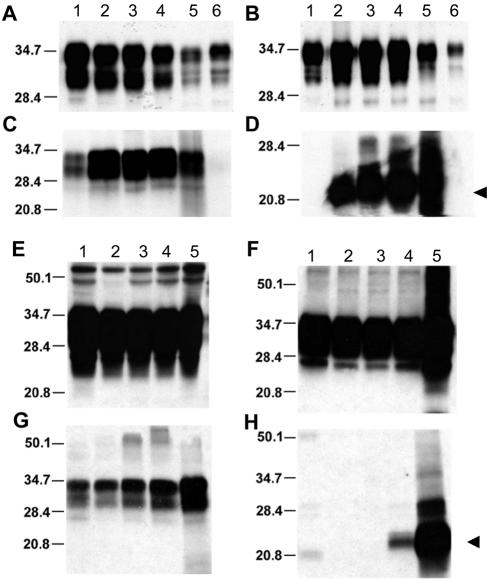
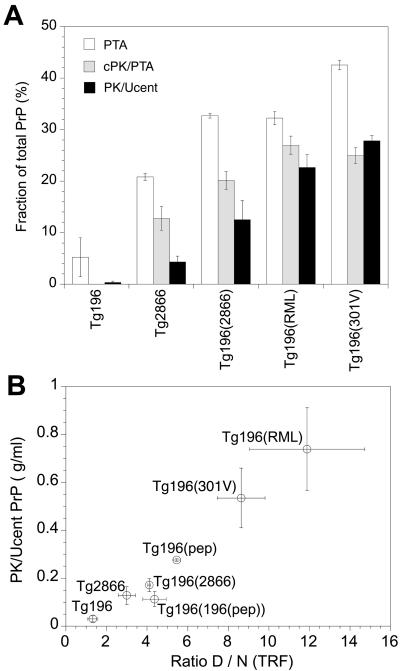
Gerstmann-Sträussler-Scheinker (GSS) disease is a dominantly inherited, human prion disease caused by a mutation in the prion protein (PrP) gene. One mutation causing GSS is P102L, denoted P101L in mouse PrP (MoPrP). In a line of transgenic mice denoted Tg2866, the P101L mutation in MoPrP produced neurodegeneration when expressed at high levels. MoPrP(Sc)(P101L) was detected both by the conformation-dependent immunoassay and after protease digestion at 4 degrees C. Transmission of prions from the brains of Tg2866 mice to those of Tg196 mice expressing low levels of MoPrP(P101L) was accompanied by accumulation of protease-resistant MoPrP(Sc)(P101L) that had previously escaped detection due to its low concentration. This conformer exhibited characteristics similar to those found in brain tissue from GSS patients. Earlier, we demonstrated that a synthetic peptide harboring the P101L mutation and folded into a beta-rich conformation initiates GSS in Tg196 mice (29). Here we report that this peptide-induced disease can be serially passaged in Tg196 mice and that the PrP conformers accompanying disease progression are conformationally indistinguishable from MoPrP(Sc)(P101L) found in Tg2866 mice developing spontaneous prion disease. In contrast to GSS prions, the 301V, RML, and 139A prion strains produced large amounts of protease-resistant PrP(Sc) in the brains of Tg196 mice. Our results argue that MoPrP(Sc)(P101L) may exist in at least several different conformations, each of which is biologically active. Such conformations occurred spontaneously in Tg2866 mice expressing high levels of MoPrP(C)(P101L) as well as in Tg196 mice expressing low levels of MoPrP(C)(P101L) that were inoculated with brain extracts from ill Tg2866 mice, with a synthetic peptide with the P101L mutation and folded into a beta-rich structure, or with prions recovered from sheep with scrapie or cattle with bovine spongiform encephalopathy.
Figures

References
-
- Baker, H. F., L. W. Duchen, J. M. Jacobs, and R. M. Ridley. 1990. Spongiform encephalopathy transmitted experimentally from Creutzfeldt-Jakob and familial Gerstmann-Sträussler-Scheinker diseases. Brain 113:1891-1909. - PubMed
-
- Barry, R. A., and S. B. Prusiner. 1986. Monoclonal antibodies to the cellular and scrapie prion proteins. J. Infect. Dis. 154:518-521. - PubMed
-
- Baskakov, I. V., C. Aagaard, I. Mehlhorn, H. Wille, D. Groth, M. A. Baldwin, S. B. Prusiner, and F. E. Cohen. 2000. Self-assembly of recombinant prion protein of 106 residues. Biochemistry 39:2792-2804. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
