

Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation
- PMID: 14749372
- PMCID: PMC344188
- DOI: 10.1128/MCB.24.4.1560-1569.2004
Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation
Abstract
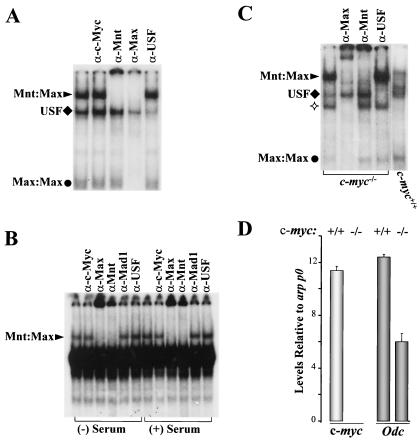
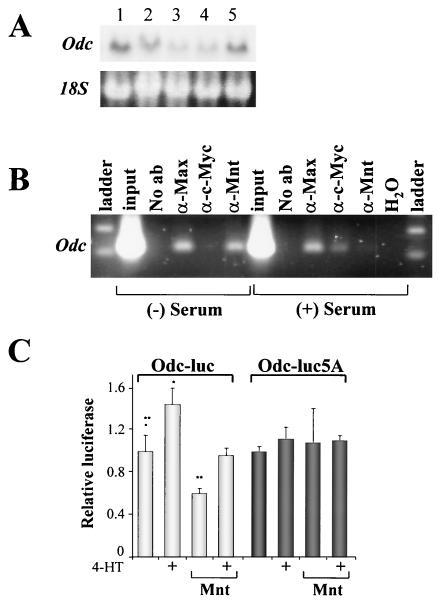
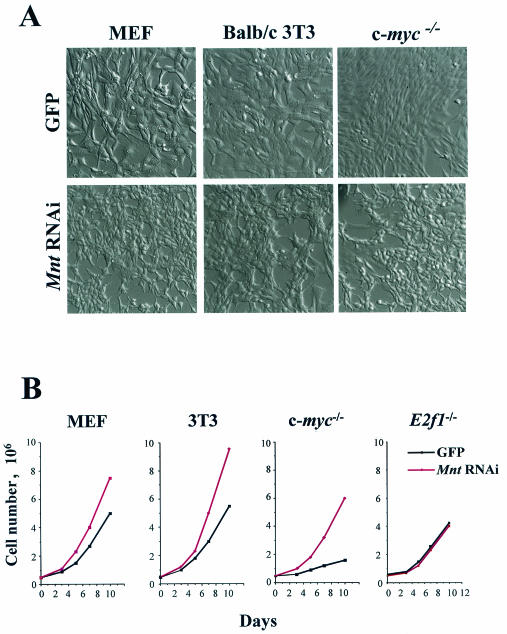
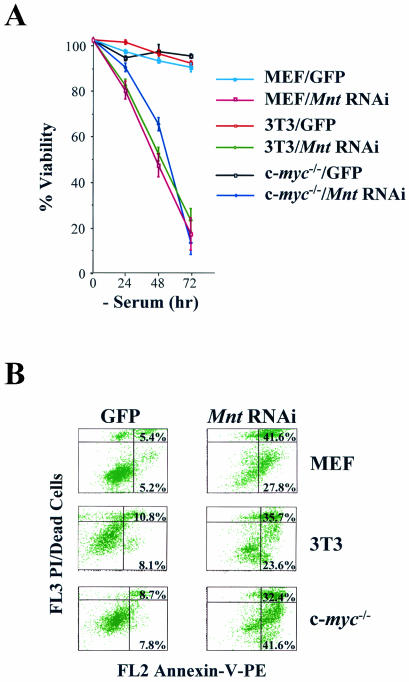
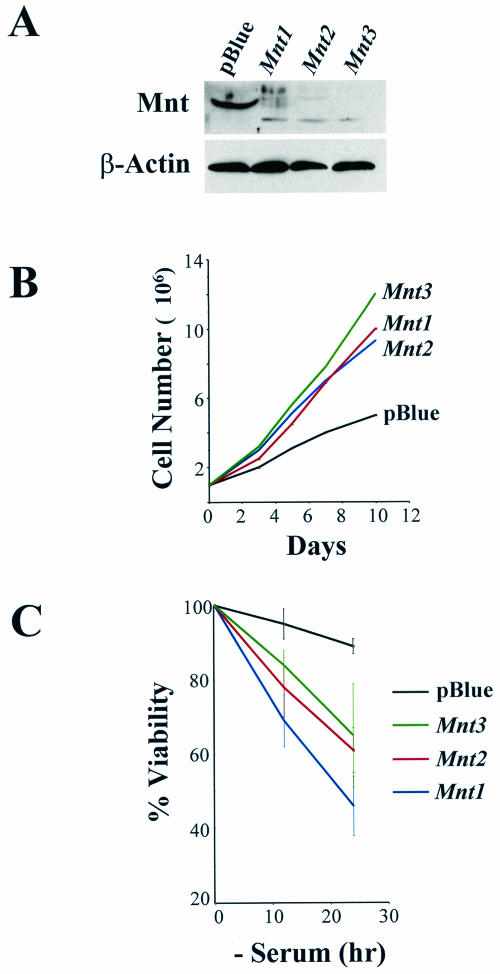
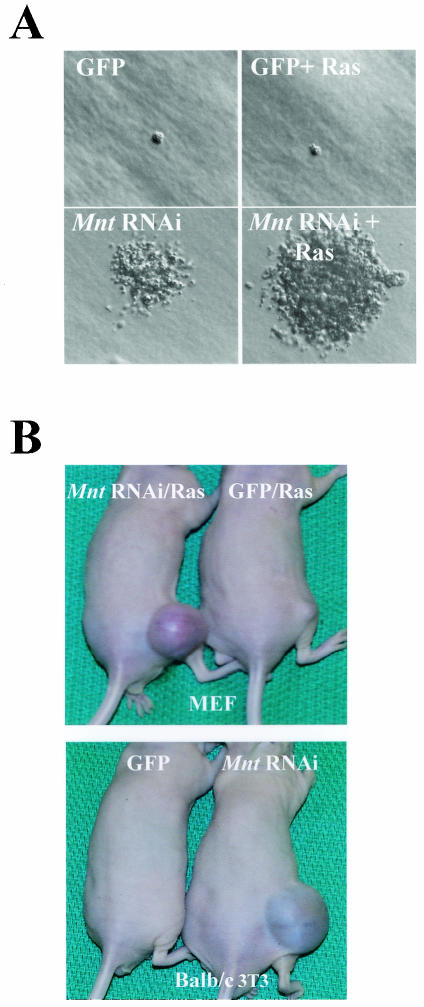
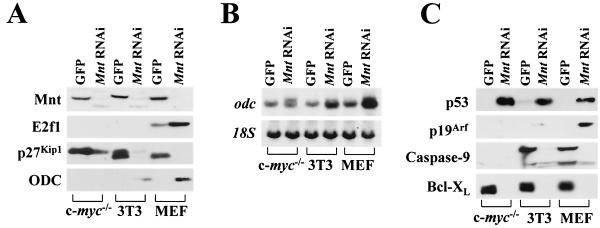
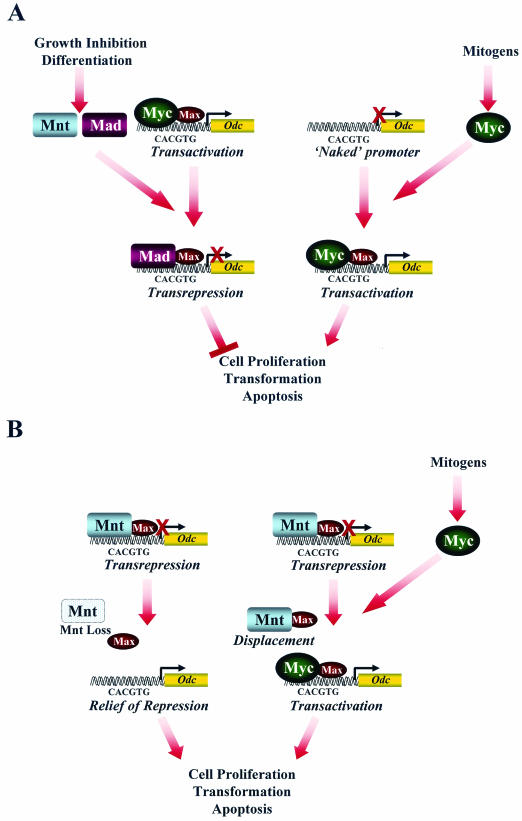
Myc oncoproteins are overexpressed in most cancers and are sufficient to accelerate cell proliferation and provoke transformation. However, in normal cells Myc also triggers apoptosis. All of the effects of Myc require its function as a transcription factor that dimerizes with Max. This complex induces genes containing CACGTG E-boxes, such as Ornithine decarboxylase (Odc), which harbors two of these elements. Here we report that in quiescent cells the Odc E-boxes are occupied by Max and Mnt, a putative Myc antagonist, and that this complex is displaced by Myc-Max complexes in proliferating cells. Knockdown of Mnt expression by stable retroviral RNA interference triggers many targets typical of the "Myc" response and provokes accelerated proliferation and apoptosis. Strikingly, these effects of Mnt knockdown are even manifest in cells lacking c-myc. Moreover, Mnt knockdown is sufficient to transform primary fibroblasts in conjunction with Ras. Therefore, Mnt behaves as a tumor suppressor. These findings support a model where Mnt represses Myc target genes and Myc functions as an oncogene by relieving Mnt-mediated repression.
Figures
References
-
- Askew, D. S., R. A. Ashmun, B. C. Simmons, and J. L. Cleveland. 1991. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 6:1915-1922. - PubMed
-
- Ayer, D. E., and R. N. Eisenman. 1993. A switch from Myc:Max to Mad:Max heterocomplexes accompanies monocyte/macrophage differentiation. Genes Dev. 7:2110-2119. - PubMed
-
- Ayer, D. E., Q. A. Lawrence, and R. N. Eisenman. 1995. Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell 80:767-776. - PubMed
-
- Baudino, T. A., K. H. Maclean, J. Brennan, E. Parganas, C. Yang, A. Aslanian, J. A. Lees, C. J. Sherr, M. F. Roussel, and J. Cleveland. 2003. Myc-mediated proliferation and lymphomagenesis, but not apoptosis, are compomised by E2f1 loss. Mol. Cell 11:1-20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials