

Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells
- PMID: 14769856
- PMCID: PMC2171976
- DOI: 10.1083/jcb.200306090
Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells
Abstract
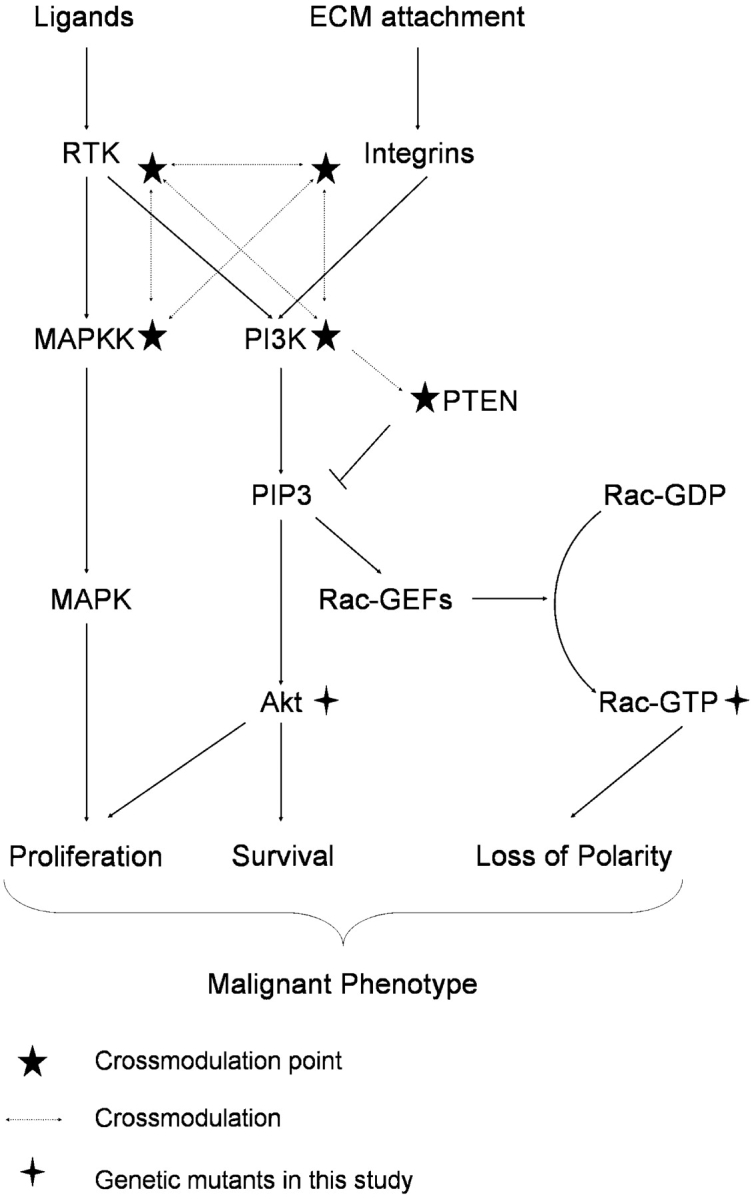
Loss of tissue polarity and increased proliferation are the characteristic alterations of the breast tumor phenotype. To investigate these processes, we used a three-dimensional (3D) culture system in which malignant human breast cells can be reverted to a normal phenotype by exposure to inhibitors of phosphatidylinositol 3-kinase (PI3K). Using this assay, we find that Akt and Rac1 act as downstream effectors of PI3K and function as control points of cellular proliferation and tissue polarity, respectively. Our results also demonstrate that the PI3K signaling pathway is an integral component of the overall signaling network induced by growth in 3D, as reversion affected by inhibition of PI3K signaling also down-modulates the endogenous levels of beta1 integrin and epidermal growth factor receptor, the upstream modulators of PI3K, and up-regulates PTEN, the antagonist of PI3K. These findings reveal key events of the PI3K pathway that play distinct roles to maintain tissue polarity and that when disrupted are instrumental in the malignant phenotype.
Copyright The Rockefeller University Press
Figures
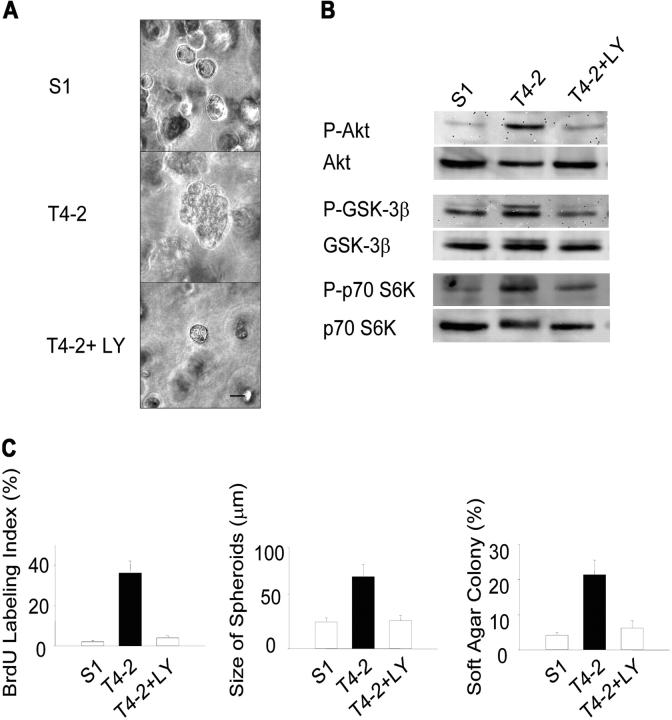
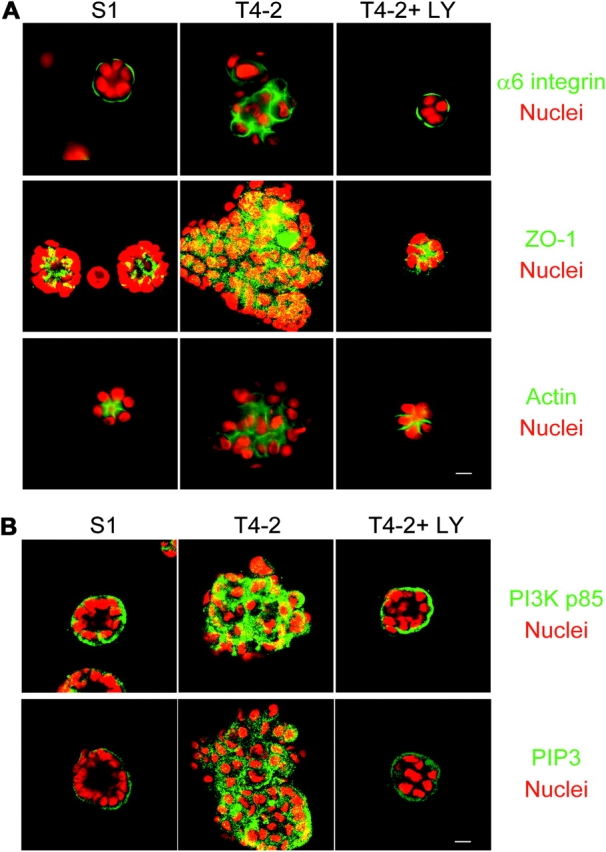
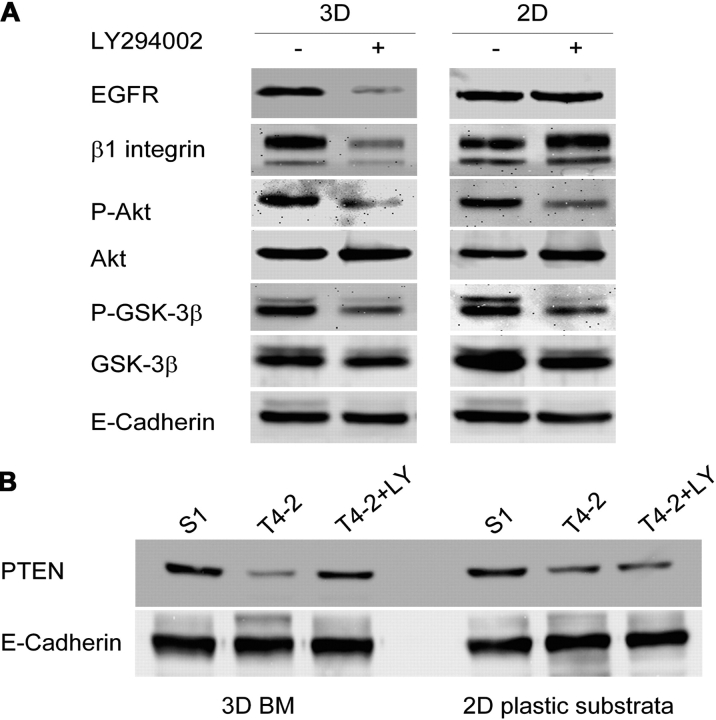

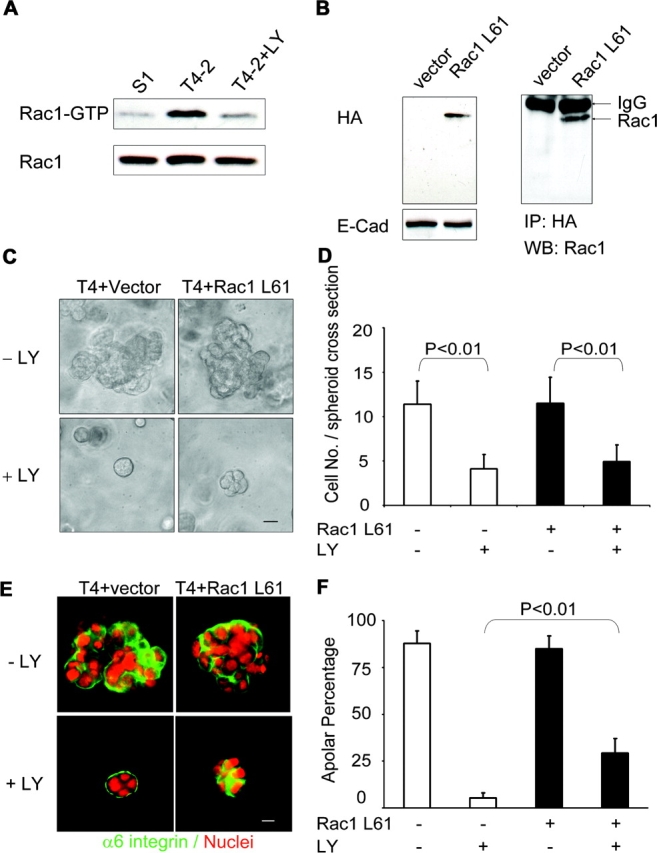

References
-
- Chan, T.O., S.E. Rittenhouse, and P.N. Tsichlis. 1999. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 68:965–1014. - PubMed
-
- Chen, R., V.H. Kang, J. Chen, J.C. Shope, J. Torabinejad, D.B. DeWald, and G.D. Prestwich. 2002. A monoclonal antibody to visualize PtdIns(3,4,5)P(3) in cells. J. Histochem. Cytochem. 50:697–708. - PubMed
-
- Comer, F.I., and C.A. Parent. 2002. PI 3-kinases and PTEN: how opposites chemoattract. Cell. 109:541–544. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
