

Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis
- PMID: 14960605
- PMCID: PMC6730331
- DOI: 10.1523/JNEUROSCI.4786-03.2004
Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis
Abstract
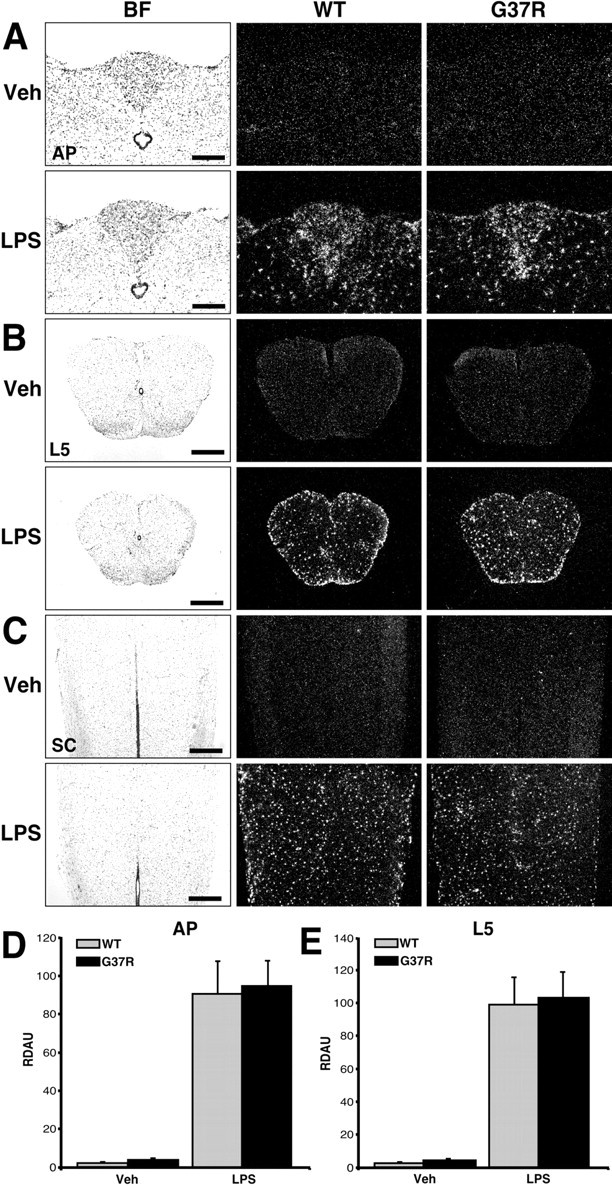
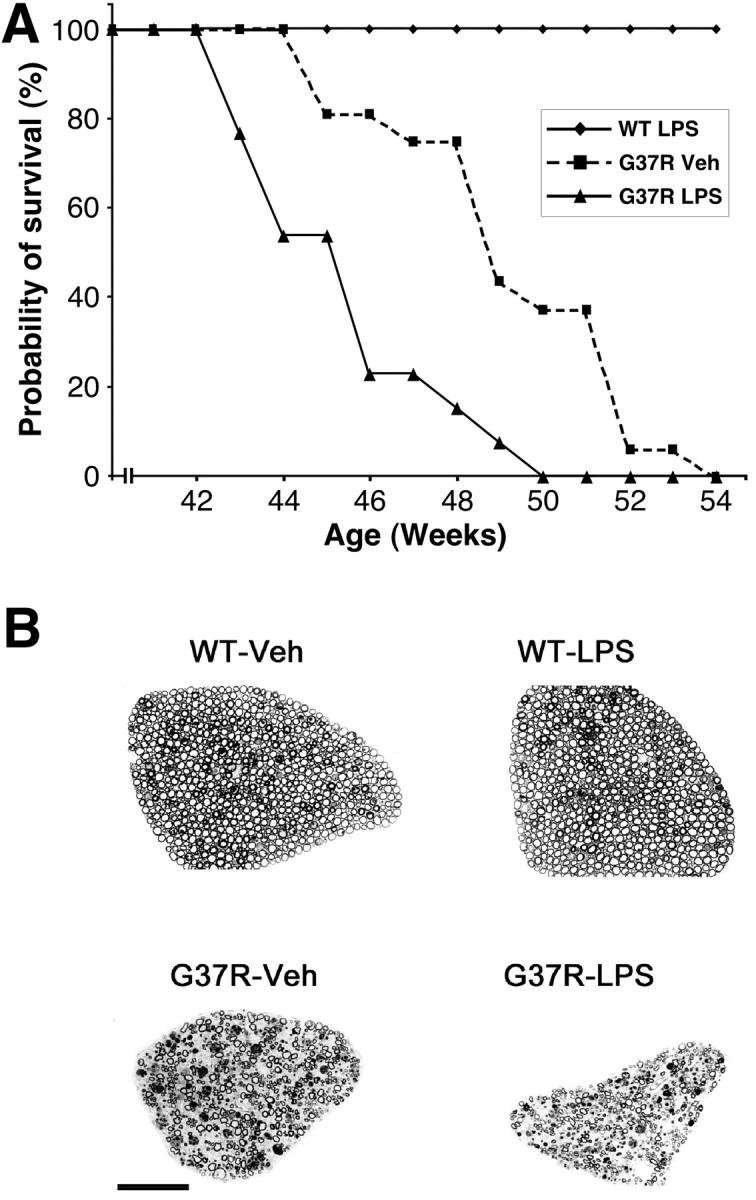
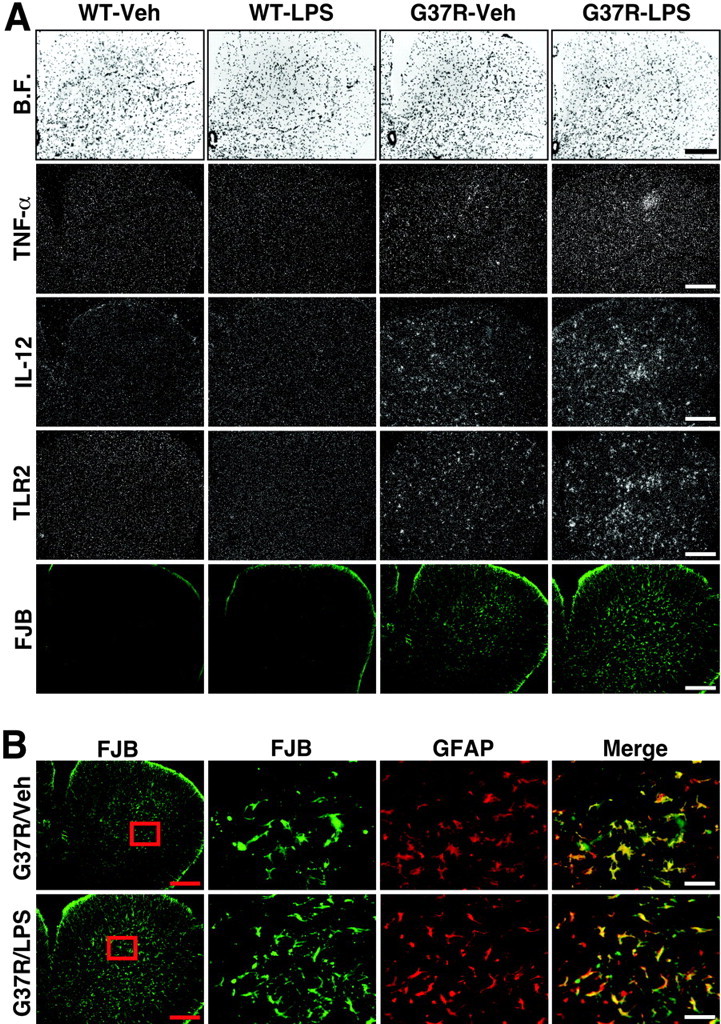
Innate immunity is a specific and organized immunological program engaged by peripheral organs and the CNS to maintain homeostasis after stress and injury. In neurodegenerative disorders, its putative deregulation, featured by inflammation and activation of glial cells resulting from inherited mutations or viral/bacterial infections, likely contributes to neuronal death. However, it remains unclear to what extent environmental factors and innate immunity cooperate to modulate the interactions between the neuronal and non-neuronal elements in the perturbed CNS. In the present study, we addressed the effects of acute and chronic administration of lipopolysaccharide (LPS), a Gram-negative bacterial wall component, in a genetic model of neurodegeneration. Transgenic mice expressing a mutant form of the superoxide dismutase 1 (SOD1(G37R)) linked to familial amyotrophic lateral sclerosis were challenged intraperitoneally with a single nontoxic or repeated injections of LPS (1 mg/kg). At different ages, SOD1(G37R) mice responded normally to acute endotoxemia. Remarkably, only a chronic challenge with LPS in presymptomatic 6-month-old SOD1(G37R) mice exacerbated disease progression by 3 weeks and motor axon degeneration. Closely associated with the severity of disease is the stronger and restricted upregulation of the receptor of innate immunity Toll-like receptor 2 and proinflammatory cytokines in degenerating regions of the ventral spinal cord and efferent fiber tracts of the brain from the LPS-treated SOD1(G37R) mice. This robust immune response was not accompanied by the establishment of acquired immunity. Our results provide solid evidence that environmental factors and innate immunity can cooperate to influence the course of disease of an inherited neuropathology.
Figures



References
-
- Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2: 675-680. - PubMed
-
- Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A (1999) Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science 285: 736-739. - PubMed
-
- Bieber AJ, Warrington A, Pease LR, Rodriguez M (2001) Humoral autoimmunity as a mediator of CNS repair. Trends Neurosci 24: S39-S44. - PubMed
-
- Bristol LA, Rothstein JD (1996) Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol 39: 676-679. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous